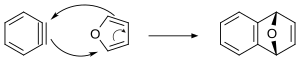In organic chemistry, an alkyne is an unsaturated hydrocarbon containing at least one carbon—carbon triple bond. The simplest acyclic alkynes with only one triple bond and no other functional groups form a homologous series with the general chemical formula CnH2n−2. Alkynes are traditionally known as acetylenes, although the name acetylene also refers specifically to C2H2, known formally as ethyne using IUPAC nomenclature. Like other hydrocarbons, alkynes are generally hydrophobic.
An ylide or ylid is a neutral dipolar molecule containing a formally negatively charged atom (usually a carbanion) directly attached to a heteroatom with a formal positive charge (usually nitrogen, phosphorus or sulfur), and in which both atoms have full octets of electrons. The result can be viewed as a structure in which two adjacent atoms are connected by both a covalent and an ionic bond; normally written X+–Y−. Ylides are thus 1,2-dipolar compounds, and a subclass of zwitterions. They appear in organic chemistry as reagents or reactive intermediates.
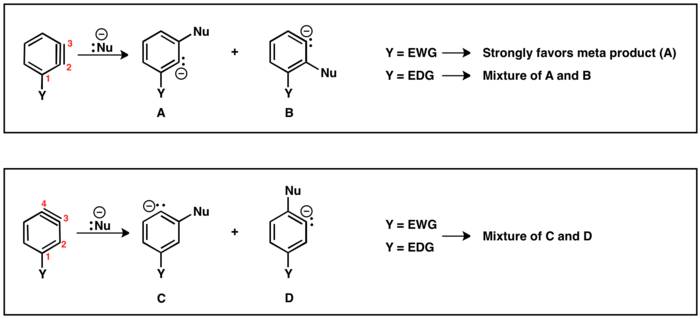
The 1,3-dipolar cycloaddition is a chemical reaction between a 1,3-dipole and a dipolarophile to form a five-membered ring. The earliest 1,3-dipolar cycloadditions were described in the late 19th century to the early 20th century, following the discovery of 1,3-dipoles. Mechanistic investigation and synthetic application were established in the 1960s, primarily through the work of Rolf Huisgen. Hence, the reaction is sometimes referred to as the Huisgen cycloaddition. 1,3-dipolar cycloaddition is an important route to the regio- and stereoselective synthesis of five-membered heterocycles and their ring-opened acyclic derivatives. The dipolarophile is typically an alkene or alkyne, but can be other pi systems. When the dipolarophile is an alkyne, aromatic rings are generally produced.
An alkyne trimerisation is a [2+2+2] cycloaddition reaction in which three alkyne units react to form a benzene ring. The reaction requires a metal catalyst. The process is of historic interest as well as being applicable to organic synthesis. Being a cycloaddition reaction, it has high atom economy. Many variations have been developed, including cyclisation of mixtures of alkynes and alkenes as well as alkynes and nitriles.
In chemical synthesis, click chemistry is a class of simple, atom-economy reactions commonly used for joining two molecular entities of choice. Click chemistry is not a single specific reaction, but describes a way of generating products that follow examples in nature, which also generates substances by joining small modular units. In many applications, click reactions join a biomolecule and a reporter molecule. Click chemistry is not limited to biological conditions: the concept of a "click" reaction has been used in chemoproteomic, pharmacological, biomimetic and molecular machinery applications. However, they have been made notably useful in the detection, localization and qualification of biomolecules.
The azide-alkyne Huisgen cycloaddition is a 1,3-dipolar cycloaddition between an azide and a terminal or internal alkyne to give a 1,2,3-triazole. Rolf Huisgen was the first to understand the scope of this organic reaction. American chemist Karl Barry Sharpless has referred to this cycloaddition as "the cream of the crop" of click chemistry and "the premier example of a click reaction".

The Bamford–Stevens reaction is a chemical reaction whereby treatment of tosylhydrazones with strong base gives alkenes. It is named for the British chemist William Randall Bamford and the Scottish chemist Thomas Stevens Stevens (1900–2000). The usage of aprotic solvents gives predominantly Z-alkenes, while protic solvent gives a mixture of E- and Z-alkenes. As an alkene-generating transformation, the Bamford–Stevens reaction has broad utility in synthetic methodology and complex molecule synthesis.

In chemistry, a phosphaalkyne is an organophosphorus compound containing a triple bond between phosphorus and carbon with the general formula R-C≡P. Phosphaalkynes are the heavier congeners of nitriles, though, due to the similar electronegativities of phosphorus and carbon, possess reactivity patterns reminiscent of alkynes. Due to their high reactivity, phosphaalkynes are not found naturally on earth, but the simplest phosphaalkyne, phosphaethyne (H-C≡P) has been observed in the interstellar medium.
In organic chemistry, a cycloalkyne is the cyclic analog of an alkyne. A cycloalkyne consists of a closed ring of carbon atoms containing one or more triple bonds. Cycloalkynes have a general formula CnH2n−4. Because of the linear nature of the C−C≡C−C alkyne unit, cycloalkynes can be highly strained and can only exist when the number of carbon atoms in the ring is great enough to provide the flexibility necessary to accommodate this geometry. Large alkyne-containing carbocycles may be virtually unstrained, while the smallest constituents of this class of molecules may experience so much strain that they have yet to be observed experimentally. Cyclooctyne is the smallest cycloalkyne capable of being isolated and stored as a stable compound. Despite this, smaller cycloalkynes can be produced and trapped through reactions with other organic molecules or through complexation to transition metals.

Organonickel chemistry is a branch of organometallic chemistry that deals with organic compounds featuring nickel-carbon bonds. They are used as a catalyst, as a building block in organic chemistry and in chemical vapor deposition. Organonickel compounds are also short-lived intermediates in organic reactions. The first organonickel compound was nickel tetracarbonyl Ni(CO)4, reported in 1890 and quickly applied in the Mond process for nickel purification. Organonickel complexes are prominent in numerous industrial processes including carbonylations, hydrocyanation, and the Shell higher olefin process.

The vinyl cation is a carbocation with the positive charge on an alkene carbon. Its empirical formula is C
2H+
3. More generally, a vinylic cation is any disubstituted carbon, where the carbon bearing the positive charge is part of a double bond and is sp hybridized. In the chemical literature, substituted vinylic cations are often referred to as vinyl cations, and understood to refer to the broad class rather than the C
2H+
3 variant alone. The vinyl cation is one of the main types of reactive intermediates involving a non-tetrahedrally coordinated carbon atom, and is necessary to explain a wide variety of observed reactivity trends. Vinyl cations are observed as reactive intermediates in solvolysis reactions, as well during electrophilic addition to alkynes, for example, through protonation of an alkyne by a strong acid. As expected from its sp hybridization, the vinyl cation prefers a linear geometry. Compounds related to the vinyl cation include allylic carbocations and benzylic carbocations, as well as aryl carbocations.

Germylenes are a class of germanium(II) compounds with the general formula :GeR2. They are heavier carbene analogs. However, unlike carbenes, whose ground state can be either singlet or triplet depending on the substituents, germylenes have exclusively a singlet ground state. Unprotected carbene analogs, including germylenes, has a dimerization nature. Free germylenes can be isolated under the stabilization of steric hindrance or electron donation. The synthesis of first stable free dialkyl germylene was reported by Jutzi, et al in 1991.
In organic chemistry, enone–alkene cycloadditions are a version of the [2+2] cycloaddition This reaction involves an enone and alkene as substrates. Although the concerted photochemical [2+2] cycloaddition is allowed, the reaction between enones and alkenes is stepwise and involves discrete diradical intermediates.
The nitrone-olefin (3+2) cycloaddition reaction is the combination of a nitrone with an alkene or alkyne to generate an isoxazoline or isoxazolidine via a (3+2) cycloaddition process. This reaction is a 1,3-dipolar cycloaddition, in which the nitrone acts as the 1,3-dipole, and the alkene or alkyne as the dipolarophile.

tert-Butylphosphaacetylene is an organophosphorus compound. Abbreviated t-BuCP, it was the first example of an isolable phosphaalkyne. Prior to its synthesis, the double bond rule had suggested that elements of Period 3 and higher were unable to form double or triple bonds with lighter main group elements because of weak orbital overlap. The synthesis of t-BuCP discredited much of the double bond rule and opened new studies into the formation of unsaturated phosphorus compounds.
The term bioorthogonal chemistry refers to any chemical reaction that can occur inside of living systems without interfering with native biochemical processes. The term was coined by Carolyn R. Bertozzi in 2003. Since its introduction, the concept of the bioorthogonal reaction has enabled the study of biomolecules such as glycans, proteins, and lipids in real time in living systems without cellular toxicity. A number of chemical ligation strategies have been developed that fulfill the requirements of bioorthogonality, including the 1,3-dipolar cycloaddition between azides and cyclooctynes, between nitrones and cyclooctynes, oxime/hydrazone formation from aldehydes and ketones, the tetrazine ligation, the isocyanide-based click reaction, and most recently, the quadricyclane ligation.
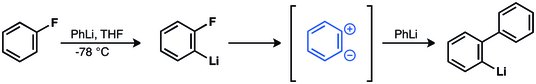
In organic chemistry, the hexadehydro-Diels–Alder (HDDA) reaction is an organic chemical reaction between a diyne and an alkyne to form a reactive benzyne species, via a [4+2] cycloaddition reaction. This benzyne intermediate then reacts with a suitable trapping agent to form a substituted aromatic product. This reaction is a derivative of the established Diels–Alder reaction and proceeds via a similar [4+2] cycloaddition mechanism. The HDDA reaction is particularly effective for forming heavily functionalized aromatic systems and multiple ring systems in one synthetic step.
Digermynes are a class of compounds that are regarded as the heavier digermanium analogues of alkynes. The parent member of this entire class is HGeGeH, which has only been characterized computationally, but has revealed key features of the whole class. Because of the large interatomic repulsion between two Ge atoms, only kinetically stabilized digermyne molecules can be synthesized and characterized by utilizing bulky protecting groups and appropriate synthetic methods, for example, reductive coupling of germanium(II) halides.

1,3-Diphenylisobenzofuran is a highly reactive diene that can scavenge unstable and short-lived dienophiles in a Diels-Alder reaction. It is furthermore used as a standard reagent for the determination of singlet oxygen, even in biological systems. Cycloadditions with 1,3-diphenylisobenzofuran and subsequent oxygen cleavage provide access to a variety of polyaromatics.
An organic azide is an organic compound that contains an azide functional group. Because of the hazards associated with their use, few azides are used commercially although they exhibit interesting reactivity for researchers. Low molecular weight azides are considered especially hazardous and are avoided. In the research laboratory, azides are precursors to amines. They are also popular for their participation in the "click reaction" between an azide and an alkyne and in Staudinger ligation. These two reactions are generally quite reliable, lending themselves to combinatorial chemistry.