
Bcl-2, encoded in humans by the BCL2 gene, is the founding member of the Bcl-2 family of regulator proteins that regulate cell death (apoptosis), by either inhibiting (anti-apoptotic) or inducing (pro-apoptotic) apoptosis. It was the first apoptosis regulator identified in any organism.

Autophagy is the natural, conserved degradation of the cell that removes unnecessary or dysfunctional components through a lysosome-dependent regulated mechanism. It allows the orderly degradation and recycling of cellular components. Although initially characterized as a primordial degradation pathway induced to protect against starvation, it has become increasingly clear that autophagy also plays a major role in the homeostasis of non-starved cells. Defects in autophagy have been linked to various human diseases, including neurodegeneration and cancer, and interest in modulating autophagy as a potential treatment for these diseases has grown rapidly.

The bafilomycins are a family of macrolide antibiotics produced from a variety of Streptomycetes. Their chemical structure is defined by a 16-membered lactone ring scaffold. Bafilomycins exhibit a wide range of biological activity, including anti-tumor, anti-parasitic, immunosuppressant and anti-fungal activity. The most used bafilomycin is bafilomycin A1, a potent inhibitor of cellular autophagy. Bafilomycins have also been found to act as ionophores, transporting potassium K+ across biological membranes and leading to mitochondrial damage and cell death.

Apoptosis regulator BAX, also known as bcl-2-like protein 4, is a protein that in humans is encoded by the BAX gene. BAX is a member of the Bcl-2 gene family. BCL2 family members form hetero- or homodimers and act as anti- or pro-apoptotic regulators that are involved in a wide variety of cellular activities. This protein forms a heterodimer with BCL2, and functions as an apoptotic activator. This protein is reported to interact with, and increase the opening of, the mitochondrial voltage-dependent anion channel (VDAC), which leads to the loss in membrane potential and the release of cytochrome c. The expression of this gene is regulated by the tumor suppressor P53 and has been shown to be involved in P53-mediated apoptosis.

The BH3 interacting-domain death agonist, or BID, gene is a pro-apoptotic member of the Bcl-2 protein family. Bcl-2 family members share one or more of the four characteristic domains of homology entitled the Bcl-2 homology (BH) domains, and can form hetero- or homodimers. Bcl-2 proteins act as anti- or pro-apoptotic regulators that are involved in a wide variety of cellular activities.

Bcl-2 homologous antagonist/killer is a protein that in humans is encoded by the BAK1 gene on chromosome 6. The protein encoded by this gene belongs to the BCL2 protein family. BCL2 family members form oligomers or heterodimers and act as anti- or pro-apoptotic regulators that are involved in a wide variety of cellular activities. This protein localizes to mitochondria, and functions to induce apoptosis. It interacts with and accelerates the opening of the mitochondrial voltage-dependent anion channel, which leads to a loss in membrane potential and the release of cytochrome c. This protein also interacts with the tumor suppressor P53 after exposure to cell stress.

The BCL2 associated agonist of cell death (BAD) protein is a pro-apoptotic member of the Bcl-2 gene family which is involved in initiating apoptosis. BAD is a member of the BH3-only family, a subfamily of the Bcl-2 family. It does not contain a C-terminal transmembrane domain for outer mitochondrial membrane and nuclear envelope targeting, unlike most other members of the Bcl-2 family. After activation, it is able to form a heterodimer with anti-apoptotic proteins and prevent them from stopping apoptosis.

Bcl-2-like protein 1 is a protein encoded in humans by the BCL2L1 gene. Through alternative splicing, the gene encodes both of the human proteins Bcl-xL and Bcl-xS.

DnaJ homolog subfamily A member 3, mitochondrial, also known as Tumorous imaginal disc 1 (TID1), is a protein that in humans is encoded by the DNAJA3 gene on chromosome 16. This protein belongs to the DNAJ/Hsp40 protein family, which is known for binding and activating Hsp70 chaperone proteins to perform protein folding, degradation, and complex assembly. As a mitochondrial protein, it is involved in maintaining membrane potential and mitochondrial DNA (mtDNA) integrity, as well as cellular processes such as cell movement, growth, and death. Furthermore, it is associated with a broad range of diseases, including neurodegenerative diseases, inflammatory diseases, and cancers.

Diablo homolog (DIABLO) is a mitochondrial protein that in humans is encoded by the DIABLO gene on chromosome 12. DIABLO is also referred to as second mitochondria-derived activator of caspases or SMAC. This protein binds inhibitor of apoptosis proteins (IAPs), thus freeing caspases to activate apoptosis. Due to its proapoptotic function, SMAC is implicated in a broad spectrum of tumors, and small molecule SMAC mimetics have been developed to enhance current cancer treatments.

Beclin-1 is a protein that in humans is encoded by the BECN1 gene. Beclin-1 is a mammalian ortholog of the yeast autophagy-related gene 6 (Atg6) and BEC-1 in the C. elegans nematode. This protein interacts with either BCL-2 or PI3k class III, playing a critical role in the regulation of both autophagy and cell death.

Autophagy related 5 (ATG5) is a protein that, in humans, is encoded by the ATG5 gene located on Chromosome 6. It is an E3 ubi autophagic cell death. ATG5 is a key protein involved in the extension of the phagophoric membrane in autophagic vesicles. It is activated by ATG7 and forms a complex with ATG12 and ATG16L1. This complex is necessary for LC3-I conjugation to PE (phosphatidylethanolamine) to form LC3-II. ATG5 can also act as a pro-apoptotic molecule targeted to the mitochondria. Under low levels of DNA damage, ATG5 can translocate to the nucleus and interact with survivin.

Mitochondrial fission 1 protein (FIS1) is a protein that in humans is encoded by the FIS1 gene on chromosome 7. This protein is a component of a mitochondrial complex, the ARCosome, that promotes mitochondrial fission. Its role in mitochondrial fission thus implicates it in the regulation of mitochondrial morphology, the cell cycle, and apoptosis. By extension, the protein is involved in associated diseases, including neurodegenerative diseases and cancers.

BCL2/adenovirus E1B 19 kDa protein-interacting protein 3-like is a protein that in humans is encoded by the BNIP3L gene.

BCL2/adenovirus E1B 19 kDa protein-interacting protein 2 is a protein that in humans is encoded by the BNIP2 gene.

Voltage-dependent anion-selective channel 1 (VDAC-1) is a beta barrel protein that in humans is encoded by the VDAC1 gene located on chromosome 5. It forms an ion channel in the outer mitochondrial membrane (OMM) and also the outer cell membrane. In the OMM, it allows ATP to diffuse out of the mitochondria into the cytoplasm. In the cell membrane, it is involved in volume regulation. Within all eukaryotic cells, mitochondria are responsible for synthesis of ATP among other metabolite needed for cell survival. VDAC1 therefore allows for communication between the mitochondrion and the cell mediating the balance between cell metabolism and cell death. Besides metabolic permeation, VDAC1 also acts as a scaffold for proteins such as hexokinase that can in turn regulate metabolism.

Autophagy related 12 is a protein that in humans is encoded by the ATG12 gene.

Bcl-2/adenovirus E1B 19 kDa-interacting protein 2-like protein is a protein that in humans is encoded by the BNIPL gene.
Mitophagy is the selective degradation of mitochondria by autophagy. It often occurs to defective mitochondria following damage or stress. The process of mitophagy was first described over a hundred years ago by Margaret Reed Lewis and Warren Harmon Lewis. Ashford and Porter used electron microscopy to observe mitochondrial fragments in liver lysosomes by 1962, and a 1977 report suggested that "mitochondria develop functional alterations which would activate autophagy." The term "mitophagy" was in use by 1998.
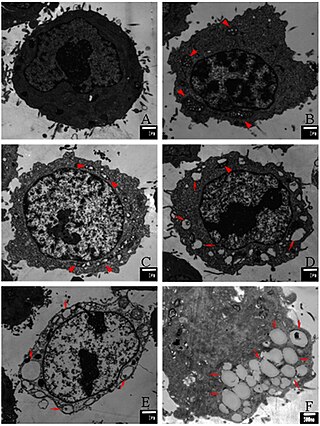
Paraptosis is a type of programmed cell death, morphologically distinct from apoptosis and necrosis. The defining features of paraptosis are cytoplasmic vacuolation, independent of caspase activation and inhibition, and lack of apoptotic morphology. Paraptosis lacks several of the hallmark characteristics of apoptosis, such as membrane blebbing, chromatin condensation, and nuclear fragmentation. Like apoptosis and other types of programmed cell death, the cell is involved in causing its own death, and gene expression is required. This is in contrast to necrosis, which is non-programmed cell death that results from injury to the cell.







