Related Research Articles

In bioinformatics, a sequence alignment is a way of arranging the sequences of DNA, RNA, or protein to identify regions of similarity that may be a consequence of functional, structural, or evolutionary relationships between the sequences. Aligned sequences of nucleotide or amino acid residues are typically represented as rows within a matrix. Gaps are inserted between the residues so that identical or similar characters are aligned in successive columns. Sequence alignments are also used for non-biological sequences, such as calculating the distance cost between strings in a natural language or in financial data.
Grammar theory to model symbol strings originated from work in computational linguistics aiming to understand the structure of natural languages. Probabilistic context free grammars (PCFGs) have been applied in probabilistic modeling of RNA structures almost 40 years after they were introduced in computational linguistics.
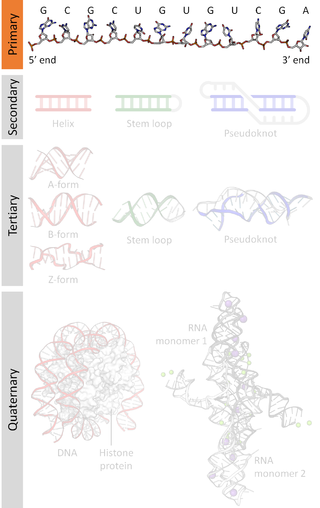
A nucleic acid sequence is a succession of bases within the nucleotides forming alleles within a DNA or RNA (GACU) molecule. This succession is denoted by a series of a set of five different letters that indicate the order of the nucleotides. By convention, sequences are usually presented from the 5' end to the 3' end. For DNA, with its double helix, there are two possible directions for the notated sequence; of these two, the sense strand is used. Because nucleic acids are normally linear (unbranched) polymers, specifying the sequence is equivalent to defining the covalent structure of the entire molecule. For this reason, the nucleic acid sequence is also termed the primary structure.

Structural alignment attempts to establish homology between two or more polymer structures based on their shape and three-dimensional conformation. This process is usually applied to protein tertiary structures but can also be used for large RNA molecules. In contrast to simple structural superposition, where at least some equivalent residues of the two structures are known, structural alignment requires no a priori knowledge of equivalent positions. Structural alignment is a valuable tool for the comparison of proteins with low sequence similarity, where evolutionary relationships between proteins cannot be easily detected by standard sequence alignment techniques. Structural alignment can therefore be used to imply evolutionary relationships between proteins that share very little common sequence. However, caution should be used in using the results as evidence for shared evolutionary ancestry because of the possible confounding effects of convergent evolution by which multiple unrelated amino acid sequences converge on a common tertiary structure.
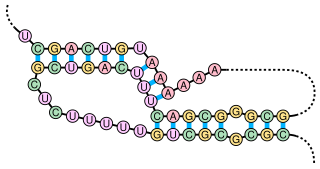
A pseudoknot is a nucleic acid secondary structure containing at least two stem-loop structures in which half of one stem is intercalated between the two halves of another stem. The pseudoknot was first recognized in the turnip yellow mosaic virus in 1982. Pseudoknots fold into knot-shaped three-dimensional conformations but are not true topological knots. These structures are categorized as cross (X) topology within the circuit topology framework, which, in contrast to knot theory, is a contact-based approach.
Pfam is a database of protein families that includes their annotations and multiple sequence alignments generated using hidden Markov models. The most recent version, Pfam 35.0, was released in November 2021 and contains 19,632 families.
Nucleic acid structure prediction is a computational method to determine secondary and tertiary nucleic acid structure from its sequence. Secondary structure can be predicted from one or several nucleic acid sequences. Tertiary structure can be predicted from the sequence, or by comparative modeling.
This list of structural comparison and alignment software is a compilation of software tools and web portals used in pairwise or multiple structural comparison and structural alignment.
Rfam is a database containing information about non-coding RNA (ncRNA) families and other structured RNA elements. It is an annotated, open access database originally developed at the Wellcome Trust Sanger Institute in collaboration with Janelia Farm, and currently hosted at the European Bioinformatics Institute. Rfam is designed to be similar to the Pfam database for annotating protein families.
Protein function prediction methods are techniques that bioinformatics researchers use to assign biological or biochemical roles to proteins. These proteins are usually ones that are poorly studied or predicted based on genomic sequence data. These predictions are often driven by data-intensive computational procedures. Information may come from nucleic acid sequence homology, gene expression profiles, protein domain structures, text mining of publications, phylogenetic profiles, phenotypic profiles, and protein-protein interaction. Protein function is a broad term: the roles of proteins range from catalysis of biochemical reactions to transport to signal transduction, and a single protein may play a role in multiple processes or cellular pathways.

In molecular biology and genetics, DNA annotation or genome annotation is the process of describing the structure and function of the components of a genome, by analyzing and interpreting them in order to extract their biological significance and understand the biological processes in which they participate. Among other things, it identifies the locations of genes and all the coding regions in a genome and determines what those genes do.
αr7 is a family of bacterial small non-coding RNAs with representatives in a broad group of Alphaproteobacterial species from the order Hyphomicrobiales. The first member of this family was found in a Sinorhizobium meliloti 1021 locus located in the chromosome (C). Further homology and structure conservation analysis identified full-length homologs in several nitrogen-fixing symbiotic rhizobia, in the plant pathogens belonging to Agrobacterium species as well as in a broad spectrum of Brucella species. αr7 RNA species are 134-159 nucleotides (nt) long and share a well defined common secondary structure. αr7 transcripts can be catalogued as trans-acting sRNAs expressed from well-defined promoter regions of independent transcription units within intergenic regions (IGRs) of the Alphaproteobacterial genomes.
αr9 is a family of bacterial small non-coding RNAs with representatives in a broad group of α-proteobacteria from the order Hyphomicrobiales. The first member of this family (Smr9C) was found in a Sinorhizobium meliloti 1021 locus located in the chromosome (C). Further homology and structure conservation analysis have identified full-length Smr9C homologs in several nitrogen-fixing symbiotic rhizobia, in the plant pathogens belonging to Agrobacterium species as well as in a broad spectrum of Brucella species. αr9C RNA species are 144-158 nt long and share a well defined common secondary structure consisting of seven conserved regions. Most of the αr9 transcripts can be catalogued as trans-acting sRNAs expressed from well-defined promoter regions of independent transcription units within intergenic regions (IGRs) of the α-proteobacterial genomes.

RNAs Associated with Genes Associated with Twister and Hammerhead ribozymes (RAGATH) refers to a bioinformatics strategy that was devised to find self-cleaving ribozymes in bacteria. It also refers to candidate RNAs, or RAGATH RNA motifs, discovered using this strategy.
SEA-PHAGES stands for Science Education Alliance-Phage Hunters Advancing Genomics and Evolutionary Science; it was formerly called the National Genomics Research Initiative. This was the first initiative launched by the Howard Hughes Medical Institute (HHMI) Science Education Alliance (SEA) by their director Tuajuanda C. Jordan in 2008 to improve the retention of Science, technology, engineering, and mathematics (STEM) students. SEA-PHAGES is a two-semester undergraduate research program administered by the University of Pittsburgh's Graham Hatfull's group and the Howard Hughes Medical Institute's Science Education Division. Students from over 100 universities nationwide engage in authentic individual research that includes a wet-bench laboratory and a bioinformatics component.

An array of protein tandem repeats is defined as several adjacent copies having the same or similar sequence motifs. These periodic sequences are generated by internal duplications in both coding and non-coding genomic sequences. Repetitive units of protein tandem repeats are considerably diverse, ranging from the repetition of a single amino acid to domains of 100 or more residues.
DIMPL is a bioinformatic pipeline that enables the extraction and selection of bacterial GC-rich intergenic regions (IGRs) that are enriched for structured non-coding RNAs (ncRNAs). The method of enriching bacterial IGRs for ncRNA motif discovery was first reported for a study in "Genome-wide discovery of structured noncoding RNAs in bacteria".
An RNA motif is a description of a group of RNAs that have a related structure. RNA motifs consist of a pattern of features within the primary sequence and secondary structure of related RNAs. Thus, it extends the concept of a sequence motif to include RNA secondary structure. The term "RNA motif" can refer both to the pattern and to the RNA sequences that match it.
References
- ↑ Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (September 1997). "Gapped BLAST and PSI-BLAST: a new generation of protein database search programs". Nucleic Acids Res. 25 (17): 3389–3402. doi:10.1093/nar/25.17.3389. PMC 146917 . PMID 9254694.
- ↑ Eddy SR, Durbin R (June 1994). "RNA sequence analysis using covariance models". Nucleic Acids Res. 22 (11): 2079–2088. doi:10.1093/nar/22.11.2079. PMC 308124 . PMID 8029015.
- ↑ Nawrocki EP, Eddy SR (November 2013). "Infernal 1.1: 100-fold faster RNA homology searches". Bioinformatics. 29 (22): 2933–2935. doi:10.1093/bioinformatics/btt509. PMC 3810854 . PMID 24008419.
- ↑ Lowe TM, Eddy SR (March 1997). "tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence". Nucleic Acids Res. 25 (5): 955–964. doi:10.1093/nar/25.5.955. PMC 146525 . PMID 9023104.
- ↑ Lowe TM, Eddy SR (February 1999). "A computational screen for methylation guide snoRNAs in yeast". Science. 283 (5405): 1168–1171. Bibcode:1999Sci...283.1168L. doi:10.1126/science.283.5405.1168. PMID 10024243. S2CID 8084145.
- ↑ Hertel J, Hofacker IL, Stadler PF (January 2008). "SnoReport: computational identification of snoRNAs with unknown targets". Bioinformatics. 24 (2): 158–164. doi: 10.1093/bioinformatics/btm464 . PMID 17895272.
- ↑ Tempel S, Tahi F (2012). "A fast ab-initio method for predicting miRNA precursors in genomes". Nucleic Acids Res. 40 (11): 955–964. doi:10.1093/nar/gks146. PMC 3367186 . PMID 22362754.
- ↑ Artzi S, Kiezun A, Shomron N (2008). "miRNAminer: a tool for homologous microRNA gene search". BMC Bioinformatics. 9 (1): 39. doi:10.1186/1471-2105-9-39. PMC 2258288 . PMID 18215311.
- 1 2 3 Rivas E, Eddy SR (2001). "Noncoding RNA gene detection using comparative sequence analysis". BMC Bioinformatics. 2: 8. doi:10.1186/1471-2105-2-8. PMC 64605 . PMID 11801179.
- ↑ Tseng HH, Weinberg Z, Gore J, Breaker RR, Ruzzo WL (April 2009). "Finding non-coding RNAs through genome-scale clustering". J Bioinform Comput Biol. 7 (2): 373–388. doi:10.1142/s0219720009004126. PMC 3417115 . PMID 19340921.
- 1 2 Weinberg Z, Barrick JE, Yao Z, Roth A, Kim JN, Gore J, Wang JX, Lee ER, Block KF, Sudarsan N, Neph S, Tompa M, Ruzzo WL, Breaker RR (2007). "Identification of 22 candidate structured RNAs in bacteria using the CMfinder comparative genomics pipeline". Nucleic Acids Res. 35 (14): 4809–4819. doi:10.1093/nar/gkm487. PMC 1950547 . PMID 17621584.
- 1 2 Hammond MC, Wachter A, Breaker RR (May 2009). "A plant 5S ribosomal RNA mimic regulates alternative splicing of transcription factor IIIA pre-mRNAs". Nat. Struct. Mol. Biol. 16 (5): 541–549. doi:10.1038/nsmb.1588. PMC 2680232 . PMID 19377483.
- ↑ Heyne S, Costa F, Rose D, Backofen R (June 2012). "GraphClust: alignment-free structural clustering of local RNA secondary structures". Bioinformatics. 28 (12): i224–32. doi:10.1093/bioinformatics/bts224. PMC 3371856 . PMID 22689765.
- ↑ Pedersen JS, Bejerano G, Siepel A, Rosenbloom K, Lindblad-Toh K, Lander ES, Kent J, Miller W, Haussler D (April 2006). "Identification and classification of conserved RNA secondary structures in the human genome". PLOS Comput. Biol. 2 (4): e33. Bibcode:2006PLSCB...2...33P. doi:10.1371/journal.pcbi.0020033. PMC 1440920 . PMID 16628248.
- ↑ Washietl S, Hofacker IL, Stadler PF (February 2005). "Fast and reliable prediction of noncoding RNAs". Proc. Natl. Acad. Sci. U.S.A. 102 (7): 2454–2459. doi: 10.1073/pnas.0409169102 . PMC 548974 . PMID 15665081.
- ↑ Weinberg Z, Wang JX, Bogue J, Yang J, Corbino K, Moy RH, Breaker RR (2010). "Comparative genomics reveals 104 candidate structured RNAs from bacteria, archaea, and their metagenomes". Genome Biol. 11 (3): R31. doi:10.1186/gb-2010-11-3-r31. PMC 2864571 . PMID 20230605.
- ↑ Weinberg Z, Lünse CE, Corbino KA, Ames TD, Nelson JW, Roth A, Perkins KR, Sherlock ME, Breaker RR (October 2017). "Detection of 224 candidate structured RNAs by comparative analysis of specific subsets of intergenic regions". Nucleic Acids Res. 45 (18): 10811–10823. doi:10.1093/nar/gkx699. PMC 5737381 . PMID 28977401.