The Protein Data Bank (PDB) is a database for the three-dimensional structural data of large biological molecules, such as proteins and nucleic acids. The data, typically obtained by X-ray crystallography, NMR spectroscopy, or, increasingly, cryo-electron microscopy, and submitted by biologists and biochemists from around the world, are freely accessible on the Internet via the websites of its member organisations. The PDB is overseen by an organization called the Worldwide Protein Data Bank, wwPDB.

In 3D computer graphics, a voxel represents a value on a regular grid in three-dimensional space. As with pixels in a 2D bitmap, voxels themselves do not typically have their position explicitly encoded with their values. Instead, rendering systems infer the position of a voxel based upon its position relative to other voxels.

Tomography is imaging by sections or sectioning that uses any kind of penetrating wave. The method is used in radiology, archaeology, biology, atmospheric science, geophysics, oceanography, plasma physics, materials science, astrophysics, quantum information, and other areas of science. The word tomography is derived from Ancient Greek τόμος tomos, "slice, section" and γράφω graphō, "to write" or, in this context as well, "to describe." A device used in tomography is called a tomograph, while the image produced is a tomogram.
Analyze is a software package developed by the Biomedical Imaging Resource (BIR) at Mayo Clinic for multi-dimensional display, processing, and measurement of multi-modality biomedical images. It is a commercial program and is used for medical tomographic scans from magnetic resonance imaging, computed tomography and positron emission tomography.

Structural bioinformatics is the branch of bioinformatics that is related to the analysis and prediction of the three-dimensional structure of biological macromolecules such as proteins, RNA, and DNA. It deals with generalizations about macromolecular 3D structures such as comparisons of overall folds and local motifs, principles of molecular folding, evolution, binding interactions, and structure/function relationships, working both from experimentally solved structures and from computational models. The term structural has the same meaning as in structural biology, and structural bioinformatics can be seen as a part of computational structural biology. The main objective of structural bioinformatics is the creation of new methods of analysing and manipulating biological macromolecular data in order to solve problems in biology and generate new knowledge.
A chemical file format is a type of data file which is used specifically for depicting molecular data. One of the most widely used is the chemical table file format, which is similar to Structure Data Format (SDF) files. They are text files that represent multiple chemical structure records and associated data fields. The XYZ file format is a simple format that usually gives the number of atoms in the first line, a comment on the second, followed by a number of lines with atomic symbols and cartesian coordinates. The Protein Data Bank Format is commonly used for proteins but is also used for other types of molecules. There are many other types which are detailed below. Various software systems are available to convert from one format to another.
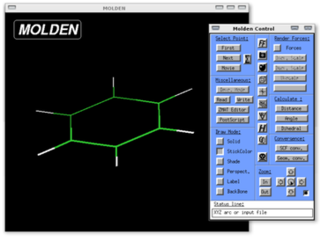
Molden is a general molecular and electronic structure processing program.
CCP4 can refer to any one of the following:
The Collaborative Computational Project Number 4 in protein crystallography (CCP4) was set up in 1979 in the United Kingdom to support collaboration between researchers working in software development and assemble a comprehensive collection of software for structural biology. The CCP4 core team is located at the Research Complex at Harwell (RCaH) at Rutherford Appleton Laboratory (RAL) in Didcot, near Oxford, UK.

UCSF Chimera is an extensible program for interactive visualization and analysis of molecular structures and related data, including density maps, supramolecular assemblies, sequence alignments, docking results, trajectories, and conformational ensembles. High-quality images and movies can be created. Chimera includes complete documentation and can be downloaded free of charge for noncommercial use.
MRC is a file format that has become an industry standard in cryo-electron microscopy (cryoEM) and electron tomography (ET), where the result of the technique is a three-dimensional grid of voxels each with a value corresponding to electron density or electric potential. It was developed by the MRC Laboratory of Molecular Biology. In 2014, the format was standardised. The format specification is available on the CCP-EM website.
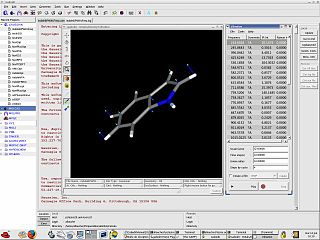
Gabedit is a graphical user interface to GAMESS (US), Gaussian, MOLCAS, MOLPRO, MPQC, OpenMopac, PC GAMESS, ORCA and Q-Chem computational chemistry packages.
Molecular replacement is a method of solving the phase problem in X-ray crystallography. MR relies upon the existence of a previously solved protein structure which is similar to our unknown structure from which the diffraction data is derived. This could come from a homologous protein, or from the lower-resolution protein NMR structure of the same protein.
Bsoft is a collection of programs and a platform for development of software for image and molecular processing in structural biology. Problems in structural biology are approached with a highly modular design, allowing fast development of new algorithms without the burden of issues such as file I/O. It provides an easily accessible interface, a resource that can be and has been used in other packages. Several workflows such as for single particle analysis and tomography are supported with parameter exchange as well as the ability to do distributed processing across heterogeneous clusters of computers.
Resolution in terms of electron density is a measure of the resolvability in the electron density map of a molecule. In X-ray crystallography, resolution is the highest resolvable peak in the diffraction pattern, while resolution in cryo-electron microscopy is a frequency space comparison of two halves of the data, which strives to correlate with the X-ray definition.
A crystallographic database is a database specifically designed to store information about the structure of molecules and crystals. Crystals are solids having, in all three dimensions of space, a regularly repeating arrangement of atoms, ions, or molecules. They are characterized by symmetry, morphology, and directionally dependent physical properties. A crystal structure describes the arrangement of atoms, ions, or molecules in a crystal.

The program Coot is used to display and manipulate atomic models of macromolecules, typically of proteins or nucleic acids, using 3D computer graphics. It is primarily focused on building and validation of atomic models into three-dimensional electron density maps obtained by X-ray crystallography methods, although it has also been applied to data from electron microscopy.
The Advanced Disc Filing System (ADFS) is a computing file system unique to the Acorn computer range and RISC OS-based successors. Initially based on the rare Acorn Winchester Filing System, it was renamed to the Advanced Disc Filing System when support for floppy discs was added and on later 32-bit systems a variant of a PC-style floppy controller.

Collaborative Computational Project Q (CCPQ) was developed in order to provide software which uses theoretical techniques to catalogue collisions between electrons, positrons or photons and atomic/molecular targets. The 'Q' stands for quantum dynamics. This project is accessible via the CCPForge website, which contains numerous other projects such as CCP2 and CCP4. The scope has increased to include atoms and molecules in strong laser fields, low-energy interactions of antihydrogen with small atoms and molecules, cold atoms, Bose–Einstein condensates and optical lattices. CCPQ gives essential information on the reactivity of various molecules, and contains two community codes R-matrix suite and MCTDH wavepacket dynamics.