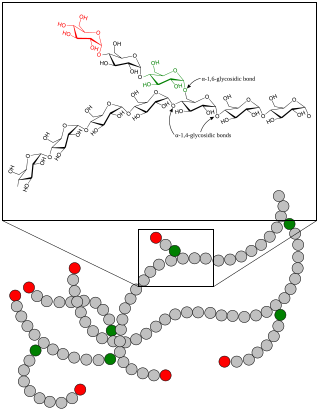
A glycogen storage disease is a metabolic disorder caused by a deficiency of an enzyme or transport protein affecting glycogen synthesis, glycogen breakdown, or glucose breakdown, typically in muscles and/or liver cells.

Limb–girdle muscular dystrophy (LGMD) is a genetically heterogeneous group of rare muscular dystrophies that share a set of clinical characteristics. It is characterised by progressive muscle wasting which affects predominantly hip and shoulder muscles. LGMD usually has an autosomal pattern of inheritance. It currently has no known cure or treatment.

Duchenne muscular dystrophy (DMD) is a severe type of muscular dystrophy predominantly affecting boys. The onset of muscle weakness typically begins around age four, with rapid progression. Initially, muscle loss occurs in the thighs and pelvis, extending to the arms, which can lead to difficulties in standing up. By the age of 12, most individuals with Duchenne muscular dystrophy are unable to walk. Affected muscles may appear larger due to an increase in fat content, and scoliosis is common. Some individuals may experience intellectual disability, and females carrying a single copy of the mutated gene may show mild symptoms.

Becker muscular dystrophy (BMD) is an X-linked recessive inherited disorder characterized by slowly progressing muscle weakness of the legs and pelvis. It is a type of dystrophinopathy. The cause is mutations and deletions in any of the 79 exons encoding the large dystrophin protein, essential for maintaining the muscle fiber's cell membrane integrity. Becker muscular dystrophy is related to Duchenne muscular dystrophy in that both result from a mutation in the dystrophin gene, however the hallmark of Becker is milder in-frame deletions. and hence has a milder course, with patients maintaining ambulation till 50–60 years if detected early.

Oculopharyngeal muscular dystrophy (OPMD) is a rare form of muscular dystrophy with symptoms generally starting when an individual is 40 to 50 years old. It can be autosomal dominant neuromuscular disease or autosomal recessive. The most common inheritance of OPMD is autosomal dominant, which means only one copy of the mutated gene needs to be present in each cell. Children of an affected parent have a 50% chance of inheriting the mutant gene.

Fukuyama congenital muscular dystrophy (FCMD) is a rare, autosomal recessive form of muscular dystrophy (weakness and breakdown of muscular tissue) mainly described in Japan but also identified in Turkish and Ashkenazi Jewish patients; fifteen cases were first described on 1960 by Dr. Yukio Fukuyama.
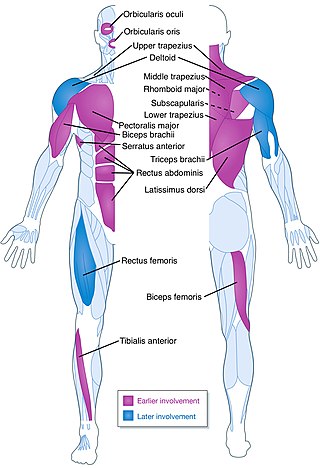
Facioscapulohumeral muscular dystrophy (FSHD) is a type of muscular dystrophy, a group of heritable diseases that cause degeneration of muscle and progressive weakness. Per the name, FSHD tends to sequentially weaken the muscles of the face, those that position the scapula, and those overlying the humerus bone of the upper arm. These areas can be spared, and muscles of other areas usually are affected, especially those of the chest, abdomen, spine, and shin. Almost any skeletal muscle can be affected in advanced disease. Abnormally positioned, termed 'winged', scapulas are common, as is the inability to lift the foot, known as foot drop. The two sides of the body are often affected unequally. Weakness typically manifests at ages 15 – 30 years. FSHD can also cause hearing loss and blood vessel abnormalities at the back of the eye.
Hereditary inclusion body myopathies (HIBM) are a group of rare genetic disorders which have different symptoms. Generally, they are neuromuscular disorders characterized by muscle weakness developing in young adults. Hereditary inclusion body myopathies comprise both autosomal recessive and autosomal dominant muscle disorders that have a variable expression (phenotype) in individuals, but all share similar structural features in the muscles.

Mitochondrial myopathies are types of myopathies associated with mitochondrial disease. Adenosine triphosphate (ATP), the chemical used to provide energy for the cell, cannot be produced sufficiently by oxidative phosphorylation when the mitochondrion is either damaged or missing necessary enzymes or transport proteins. With ATP production deficient in mitochondria, there is an over-reliance on anaerobic glycolysis which leads to lactic acidosis either at rest or exercise-induced.

Dysferlin also known as dystrophy-associated fer-1-like protein is a protein that in humans is encoded by the DYSF gene. Dysferlin is linked with plasma membrane repair., stabilization of calcium signaling and the development of the T-tubule system of the muscle A defect in the DYSF gene, located on chromosome 2p12-14, results in several types of muscular dystrophy; including Miyoshi myopathy (MM), Limb-girdle muscular dystrophy type 2B (LGMD2B) and Distal Myopathy (DM). A reduction or absence of dysferlin, termed dysferlinopathy, usually becomes apparent in the third or fourth decade of life and is characterised by weakness and wasting of various voluntary skeletal muscles. Pathogenic mutations leading to dysferlinopathy can occur throughout the DYSF gene.
Congenital muscular dystrophies are autosomal recessively-inherited muscle diseases. They are a group of heterogeneous disorders characterized by muscle weakness which is present at birth and the different changes on muscle biopsy that ranges from myopathic to overtly dystrophic due to the age at which the biopsy takes place.

Emery–Dreifuss muscular dystrophy (EDMD) is a type of muscular dystrophy, a group of heritable diseases that cause progressive impairment of muscles. EDMD affects muscles used for movement, causing atrophy, weakness and contractures. It almost always affects the heart, causing abnormal rhythms, heart failure, or sudden cardiac death. It is rare, affecting 0.39 per 100,000 people. It is named after Alan Eglin H. Emery and Fritz E. Dreifuss.
Bethlem myopathy is predominantly an autosomal dominant myopathy, classified as a congenital form of limb-girdle muscular dystrophy. There are two types of Bethlem myopathy, based on which type of collagen is affected.
Calpain-3 is a protein that in humans is encoded by the CAPN3 gene.

Beta-sarcoglycan is a protein that in humans is encoded by the SGCB gene.

Delta-sarcoglycan is a protein that in humans is encoded by the SGCD gene.

Gamma-sarcoglycan is a protein that in humans is encoded by the SGCG gene. The α to δ-sarcoglycans are expressed predominantly (β) or exclusively in striated muscle. A mutation in any of the sarcoglycan genes may lead to a secondary deficiency of the other sarcoglycan proteins, presumably due to destabilisation of the sarcoglycan complex. The disease-causing mutations in the α to δ genes cause disruptions within the dystrophin-associated protein (DAP) complex in the muscle cell membrane. The transmembrane components of the DAP complex link the cytoskeleton to the extracellular matrix in adult muscle fibres, and are essential for the preservation of the integrity of the muscle cell membrane.
Ullrich congenital muscular dystrophy (UCMD) is a form of congenital muscular dystrophy. There are two forms: UCMD1 and UCMD2.
Multi/minicore myopathy is a congenital myopathy usually caused by mutations in either the SELENON and RYR1 genes. It is characterised the presence of multifocal, well-circumscribed areas with reduction of oxidative staining and low myofibrillar ATPase on muscle biopsy. It is also known as Minicore myopathy, Multicore myopathy, Multiminicore myopathy, Minicore myopathy with external ophthalmoplegia, Multicore myopathy with external ophthalmoplegia and Multiminicore disease with external ophthalmoplegia.

Anoctamin 5 (ANO5) is a protein that in humans is encoded by the ANO5 gene.



