
Carnitine is a quaternary ammonium compound involved in metabolism in most mammals, plants, and some bacteria. In support of energy metabolism, carnitine transports long-chain fatty acids from the cytosol into mitochondria to be oxidized for free energy production, and also participates in removing products of metabolism from cells. Given its key metabolic roles, carnitine is concentrated in tissues like skeletal and cardiac muscle that metabolize fatty acids as an energy source. Generally individuals, including strict vegetarians, synthesize enough L-carnitine in vivo.

Medium-chain acyl-CoA dehydrogenase deficiency is a disorder of fatty acid oxidation that impairs the body's ability to break down medium-chain fatty acids into acetyl-CoA. The disorder is characterized by hypoglycemia and sudden death without timely intervention, most often brought on by periods of fasting or vomiting.

Long-chain 3-hydroxyacyl-coenzyme A dehydrogenase deficiency is a rare autosomal recessive fatty acid oxidation disorder that prevents the body from converting certain fats into energy. This can become life-threatening, particularly during periods of fasting.

Systemic primary carnitine deficiency (SPCD) is an inborn error of fatty acid transport caused by a defect in the transporter responsible for moving carnitine across the plasma membrane. Carnitine is an important amino acid for fatty acid metabolism. When carnitine cannot be transported into tissues, fatty acid oxidation is impaired, leading to a variety of symptoms such as chronic muscle weakness, cardiomyopathy, hypoglycemia and liver dysfunction. The specific transporter involved with SPCD is OCTN2, coded for by the SLC22A5 gene located on chromosome 5. SPCD is inherited in an autosomal recessive manner, with mutated alleles coming from both parents.
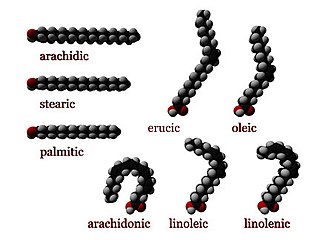
Numerous genetic disorders are caused by errors in fatty acid metabolism. These disorders may be described as fatty oxidation disorders or as a lipid storage disorders, and are any one of several inborn errors of metabolism that result from enzyme defects affecting the ability of the body to oxidize fatty acids in order to produce energy within muscles, liver, and other cell types.
Carnitine palmitoyltransferase I deficiency is a rare metabolic disorder that prevents the body from converting certain fats called long-chain fatty acids(LCFA) into energy, particularly during periods without food. It is caused by a mutation in CPT1A on chromosome 11.
Carnitine palmitoyltransferase II deficiency, sometimes shortened to CPT-II or CPT2, is an autosomal recessively inherited genetic metabolic disorder characterized by an enzymatic defect that prevents long-chain fatty acids from being transported into the mitochondria for utilization as an energy source. The disorder presents in one of three clinical forms: lethal neonatal, severe infantile hepatocardiomuscular and myopathic.
Mitochondrial trifunctional protein deficiency is an autosomal recessive fatty acid oxidation disorder that prevents the body from converting certain fats to energy, particularly during periods without food. People with this disorder have inadequate levels of an enzyme that breaks down a certain group of fats called long-chain fatty acids.
Very long-chain acyl-coenzyme A dehydrogenase deficiency is a fatty-acid metabolism disorder which prevents the body from converting certain fats to energy, particularly during periods without food.
Glutaric acidemia type 2 is an autosomal recessive metabolic disorder that is characterised by defects in the ability of the body to use proteins and fats for energy. Incompletely processed proteins and fats can build up, leading to a dangerous chemical imbalance called acidosis.

Malonyl-CoA decarboxylase deficiency (MCD) is an autosomal-recessive metabolic disorder caused by a genetic mutation that disrupts the activity of Malonyl-CoA decarboxylase. This enzyme breaks down Malonyl-CoA into acetyl-CoA and carbon dioxide.
Short-chain acyl-coenzyme A dehydrogenase deficiency (SCADD) is an autosomal recessive fatty acid oxidation disorder which affects enzymes required to break down a certain group of fats called short chain fatty acids.

Palmitoylcarnitine is an ester derivative of carnitine involved in the metabolism of fatty acids. During the tricarboxylic acid cycle (TCA), fatty acids undergo a process known as β-oxidation to produce energy in the form of ATP. β-oxidation occurs primarily within mitochondria, however the mitochondrial membrane prevents the entry of long chain fatty acids (>C10), so the conversion of fatty acids such as palmitic acid is key. Palmitic acid is brought to the cell and once inside the cytoplasm is first converted to Palmitoyl-CoA. Palmitoyl-CoA has the ability to freely pass the outer mitochondrial membrane, but the inner membrane is impermeable to the Acyl-CoA and thioester forms of various long-chain fatty acids such as palmitic acid. The palmitoyl-CoA is then enzymatically transformed into palmitoylcarnitine via the Carnitine O-palmitoyltransferase family. The palmitoylcarnitine is then actively transferred into the inner membrane of the mitochondria via the carnitine-acylcarnitine translocase. Once inside the inner mitochondrial membrane, the same Carnitine O-palmitoyltransferase family is then responsible for transforming the palmitoylcarnitine back to the palmitoyl-CoA form.
Isobutyryl-coenzyme A dehydrogenase deficiency is a rare metabolic disorder in which the body is unable to process certain amino acids properly.

Ornithine translocase deficiency, also called hyperornithinemia-hyperammonemia-homocitrullinuria (HHH) syndrome, is a rare autosomal recessive urea cycle disorder affecting the enzyme ornithine translocase, which causes ammonia to accumulate in the blood, a condition called hyperammonemia.
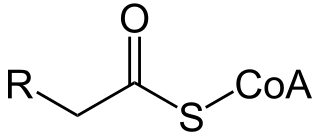
Acyl-CoA is a group of coenzymes that metabolize fatty acids. Acyl-CoA's are susceptible to beta oxidation, forming, ultimately, acetyl-CoA. The acetyl-CoA enters the citric acid cycle, eventually forming several equivalents of ATP. In this way, fats are converted to ATP, the universal biochemical energy carrier.
3-hydroxyacyl-coenzyme A dehydrogenase deficiency is a rare condition that prevents the body from converting certain fats to energy, particularly during fasting. Normally, through a process called fatty acid oxidation, several enzymes work in a step-wise fashion to metabolize fats and convert them to energy. People with 3-hydroxyacyl-coenzyme A dehydrogenase deficiency have inadequate levels of an enzyme required for a step that metabolizes groups of fats called medium chain fatty acids and short chain fatty acids; for this reason this disorder is sometimes called medium- and short-chain 3-hydroxyacyl-coenzyme A dehydrogenase (M/SCHAD) deficiency.

Carnitine palmitoyltransferase I (CPT1) also known as carnitine acyltransferase I, CPTI, CAT1, CoA:carnitine acyl transferase (CCAT), or palmitoylCoA transferase I, is a mitochondrial enzyme responsible for the formation of acyl carnitines by catalyzing the transfer of the acyl group of a long-chain fatty acyl-CoA from coenzyme A to l-carnitine. The product is often Palmitoylcarnitine, but other fatty acids may also be substrates. It is part of a family of enzymes called carnitine acyltransferases. This "preparation" allows for subsequent movement of the acyl carnitine from the cytosol into the intermembrane space of mitochondria.
A broad classification for genetic disorders that result from an inability of the body to produce or utilize an enzyme or transport protein that is required to oxidize fatty acids. They are an inborn error of lipid metabolism, and when it affects the muscles also a metabolic myopathy.

