
Half-life is the time required for a quantity to reduce to half of its initial value. The term is commonly used in nuclear physics to describe how quickly unstable atoms undergo radioactive decay or how long stable atoms survive. The term is also used more generally to characterize any type of exponential decay. For example, the medical sciences refer to the biological half-life of drugs and other chemicals in the human body. The converse of half-life is doubling time.
In classical mechanics, a harmonic oscillator is a system that, when displaced from its equilibrium position, experiences a restoring force F proportional to the displacement x:
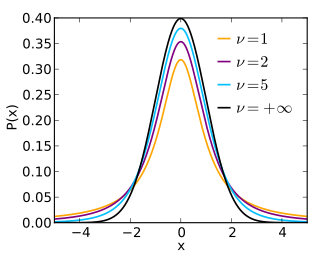
In probability and statistics, Student's t-distribution is a continuous probability distribution that generalizes the standard normal distribution. Like the latter, it is symmetric around zero and bell-shaped.
Hemorheology, also spelled haemorheology, or blood rheology, is the study of flow properties of blood and its elements of plasma and cells. Proper tissue perfusion can occur only when blood's rheological properties are within certain levels. Alterations of these properties play significant roles in disease processes. Blood viscosity is determined by plasma viscosity, hematocrit and mechanical properties of red blood cells. Red blood cells have unique mechanical behavior, which can be discussed under the terms erythrocyte deformability and erythrocyte aggregation. Because of that, blood behaves as a non-Newtonian fluid. As such, the viscosity of blood varies with shear rate. Blood becomes less viscous at high shear rates like those experienced with increased flow such as during exercise or in peak-systole. Therefore, blood is a shear-thinning fluid. Contrarily, blood viscosity increases when shear rate goes down with increased vessel diameters or with low flow, such as downstream from an obstruction or in diastole. Blood viscosity also increases with increases in red cell aggregability.

The step response of a system in a given initial state consists of the time evolution of its outputs when its control inputs are Heaviside step functions. In electronic engineering and control theory, step response is the time behaviour of the outputs of a general system when its inputs change from zero to one in a very short time. The concept can be extended to the abstract mathematical notion of a dynamical system using an evolution parameter.
In pharmacology, bioavailability is a subcategory of absorption and is the fraction (%) of an administered drug that reaches the systemic circulation.
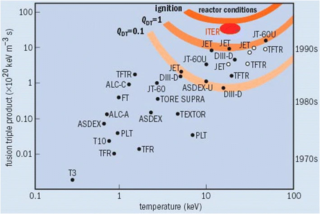
The Lawson criterion is a figure of merit used in nuclear fusion research. It compares the rate of energy being generated by fusion reactions within the fusion fuel to the rate of energy losses to the environment. When the rate of production is higher than the rate of loss, the system will produce net energy. If enough of that energy is captured by the fuel, the system will become self-sustaining and is said to be ignited.

Physiologically based pharmacokinetic (PBPK) modeling is a mathematical modeling technique for predicting the absorption, distribution, metabolism and excretion (ADME) of synthetic or natural chemical substances in humans and other animal species. PBPK modeling is used in pharmaceutical research and drug development, and in health risk assessment for cosmetics or general chemicals.
In pharmacology, clearance is a pharmacokinetic parameter representing the efficiency of drug elimination. This is the rate of elimination of a substance divided by its concentration. The parameter also indicates the theoretical volume of plasma from which a substance would be completely removed per unit time. Usually, clearance is measured in L/h or mL/min. The quantity reflects the rate of drug elimination divided by plasma concentration. Excretion, on the other hand, is a measurement of the amount of a substance removed from the body per unit time. While clearance and excretion of a substance are related, they are not the same thing. The concept of clearance was described by Thomas Addis, a graduate of the University of Edinburgh Medical School.
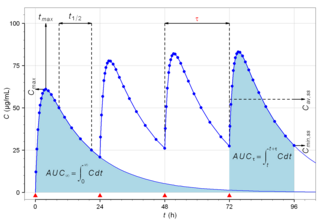
In medicine and pharmacology, a trough level or trough concentration (Ctrough) is the concentration reached by a drug immediately before the next dose is administered, often used in therapeutic drug monitoring. The name comes from the idea that on a graph of concentration versus time, the line forms a U-shaped trough at the lowest region, before a new dose sends it higher again. The usual criterion is concentration in the blood serum, although in some instances local concentration within tissues is relevant. It is pharmacokinetically normal that over time, the drug molecules are being metabolized or cleared by the body, so the concentration of drug that remains available is dropping. In a medicine that is administered periodically, the trough level should be measured just before the administration of the next dose in order to avoid overdosing. A trough level is contrasted with a "peak level" (Cmax), which is the highest level of the medicine in the body, and the "average level", which is the mean level over time. It is widely used in clinical trials for newer medicines to investigate therapeutic effectiveness and safety.
Biological half-life is the time taken for concentration of a biological substance to decrease from its maximum concentration (Cmax) to half of Cmax in the blood plasma. It is denoted by the abbreviation .
The Mason–Weaver equation describes the sedimentation and diffusion of solutes under a uniform force, usually a gravitational field. Assuming that the gravitational field is aligned in the z direction, the Mason–Weaver equation may be written
Pharmacokinetics, sometimes abbreviated as PK, is a branch of pharmacology dedicated to describing how the body affects a specific substance after administration. The substances of interest include any chemical xenobiotic such as pharmaceutical drugs, pesticides, food additives, cosmetics, etc. It attempts to analyze chemical metabolism and to discover the fate of a chemical from the moment that it is administered up to the point at which it is completely eliminated from the body. Pharmacokinetics is based on mathematical modeling that places great emphasis on the relationship between drug plasma concentration and the time elapsed since the drug's administration. Pharmacokinetics is the study of how an organism affects the drug, whereas pharmacodynamics (PD) is the study of how the drug affects the organism. Both together influence dosing, benefit, and adverse effects, as seen in PK/PD models.
The elimination rate constantK or Ke is a value used in pharmacokinetics to describe the rate at which a drug is removed from the human system.
Cmax is the maximum serum concentration that a drug achieves in a specified compartment or test area of the body after the drug has been administered and before the administration of a second dose. It is a standard measurement in pharmacokinetics.
In the field of pharmacokinetics, the area under the curve (AUC) is the definite integral of the concentration of a drug in blood plasma as a function of time. In practice, the drug concentration is measured at certain discrete points in time and the trapezoidal rule is used to estimate AUC. In pharmacology, the area under the plot of plasma concentration of a drug versus time after dosage gives insight into the extent of exposure to a drug and its clearance rate from the body.
A Logan plot is a graphical analysis technique based on the compartment model that uses linear regression to analyze pharmacokinetics of tracers involving reversible uptake. It is mainly used for the evaluation of nuclear medicine imaging data after the injection of a labeled ligand that binds reversibly to specific receptor or enzyme.
The residence time of a fluid parcel is the total time that the parcel has spent inside a control volume (e.g.: a chemical reactor, a lake, a human body). The residence time of a set of parcels is quantified in terms of the frequency distribution of the residence time in the set, which is known as residence time distribution (RTD), or in terms of its average, known as mean residence time.
In pharmacokinetics, the drug accumulation ratio (Rac) is the ratio of accumulation of a drug under steady state conditions as compared to a single dose. The higher the value, the more the drug accumulates in the body. An Rac of 1 means no accumulation.
Cavg is the average concentration of a drug in the central circulation during a dosing interval in steady state. It is calculated by







