Figures

DDX3X syndrome is a genetic disorder that affects predominantly females. Patients with DDX3X syndrome may develop developmental delay or intellectual disability, autism, ADHD, and low muscle tone. The syndrome develops due to mutations of the DDX3X gene located on the X chromosome, and the clinical picture varies depending on the specific mutation.
The syndrome was first described in a study published in 2015. [1]
According to studies performed up to 2022, DDX3X syndrome was presumed to account for 1-3% of cases of unexplained developmental delay and/or intellectual disability in females, [2] but this may be an approximate early estimate.
Neurodevelopmental disorders are a group of conditions that begin to produce symptoms during childhood. According to the American Psychiatric Association Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition, (DSM-5) published in 2013, these conditions generally appear in early childhood, usually before children start school, and can persist into adulthood. The key characteristic of all these disorders is that they negatively impact a person's functioning in one or more domains of life depending on the disorder and deficits it has caused. All of these disorders and their levels of impairment exist on a spectrum, and affected individuals can experience varying degrees of symptoms and deficits, despite having the same diagnosis.

The heritability of autism is the proportion of differences in expression of autism that can be explained by genetic variation; if the heritability of a condition is high, then the condition is considered to be primarily genetic. Autism has a strong genetic basis. Although the genetics of autism are complex, autism spectrum disorder (ASD) is explained more by multigene effects than by rare mutations with large effects.

22q13 deletion syndrome, also known as Phelan–McDermid syndrome (PMS), is a genetic disorder caused by deletions or rearrangements on the q terminal end of chromosome 22. Any abnormal genetic variation in the q13 region that presents with significant manifestations (phenotype) typical of a terminal deletion may be diagnosed as 22q13 deletion syndrome. There is disagreement among researchers as to the exact definition of 22q13 deletion syndrome. The Developmental Synaptopathies Consortium defines PMS as being caused by SHANK3 mutations, a definition that appears to exclude terminal deletions. The requirement to include SHANK3 in the definition is supported by many but not by those who first described 22q13 deletion syndrome.

Chromodomain-helicase-DNA-binding protein 2 is an enzyme that in humans is encoded by the CHD2 gene.

Intellectual disability (ID), also known as general learning disability in the United Kingdom and formerly mental retardation, is a generalized neurodevelopmental disorder characterized by significantly impaired intellectual and adaptive functioning. It is defined by an IQ under 70, in addition to deficits in two or more adaptive behaviors that affect everyday, general living. Intellectual functions are defined under DSM-V as reasoning, problem‑solving, planning, abstract thinking, judgment, academic learning, and learning from instruction and experience, and practical understanding confirmed by both clinical assessment and standardized tests. Adaptive behavior is defined in terms of conceptual, social, and practical skills involving tasks performed by people in their everyday lives.
X-linked intellectual disability refers to medical disorders associated with X-linked recessive inheritance that result in intellectual disability.

Classic autism, also known as childhood autism, autistic disorder, (early) infantile autism, infantile psychosis, Kanner's autism,Kanner's syndrome, or (formerly) just autism, is a neurodevelopmental condition first described by Leo Kanner in 1943. It is characterized by atypical and impaired development in social interaction and communication as well as restricted, repetitive behaviors, activities, and interests. These symptoms first appear in early childhood and persist throughout life.

Lujan–Fryns syndrome (LFS) is an X-linked genetic disorder that causes mild to moderate intellectual disability and features described as Marfanoid habitus, referring to a group of physical characteristics similar to those found in Marfan syndrome. These features include a tall, thin stature and long, slender limbs. LFS is also associated with psychopathology and behavioral abnormalities, and it exhibits a number of malformations affecting the brain and heart. The disorder is inherited in an X-linked dominant manner, and is attributed to a missense mutation in the MED12 gene. There is currently no treatment or therapy for the underlying MED12 malfunction, and the exact cause of the disorder remains unclear.

Pitt–Hopkins syndrome (PTHS) is a rare genetic disorder characterized by developmental delay, epilepsy, distinctive facial features, and possible intermittent hyperventilation followed by apnea. Pitt-Hopkins syndrome can be marked by intellectual disabilities as well as problems with socializing. It is part of the clinical spectrum of Rett-like syndromes.

Autism, formally called autism spectrum disorder (ASD) or autism spectrum condition (ASC), is a neurodevelopmental disorder characterized by deficits in social communication and social interaction, and repetitive or restricted patterns of behaviors, interests, or activities, which can include hyper- and hyporeactivity to sensory input. Autism is clinically regarded as a spectrum disorder, meaning that it can manifest very differently in each person. For example, some are nonspeaking, while others have proficient spoken language. Because of this, there is wide variation in the support needs of people across the autism spectrum.

SET binding protein 1 is a protein that in humans is encoded by the SETBP1 gene.

Sotos syndrome is a rare genetic disorder characterized by excessive physical growth during the first years of life. Excessive growth often starts in infancy and continues into the early teen years. The disorder may be accompanied by autism, mild intellectual disability, delayed motor, cognitive, and social development, hypotonia, and speech impairments. Children with Sotos syndrome tend to be large at birth and are often taller, heavier, and have relatively large skulls (macrocephaly) than is normal for their age. Signs of the disorder, which vary among individuals, include a disproportionately large skull with a slightly protrusive forehead, large hands and feet, large mandible, hypertelorism, and downslanting eyes. Clumsiness, an awkward gait, and unusual aggressiveness or irritability may also occur.
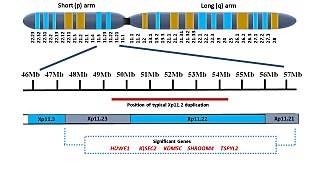
Xp11.2 duplication is a genomic variation marked by the duplication of an X chromosome region on the short arm p at position 11.2, defined by standard karyotyping (G-banding). This gene-rich, rearrangement prone region can be further divided into three loci - Xp11.21, Xp11.22 and Xp11.23. The duplication could involve any combination of these three loci. While the length of the duplication can vary from 0.5Mb to 55 Mb, most duplications measure about 4.5Mb and typically occur in the region of 11.22-11.23. Most affected females show preferential activation of the duplicated X chromosome. Features of affected individuals vary significantly, even among members of the same family. The Xp11.2 duplication can be 'silent' - presenting no obvious symptoms in carriers - which is known from the asymptomatic parents of affected children carrying the duplication. The common symptoms include intellectual disabilities, speech delay and learning difficulties, while in rare cases, children have seizures and a recognizable brain wave pattern when assessed by EEG (electroencephalography).
ADNP syndrome, also known as Helsmoortel-Van der Aa syndrome (HVDAS), is a non-inherited neurodevelopmental disorder caused by mutations in the activity-dependent neuroprotector homeobox (ADNP) gene.

CDK13-related disorder, also known as congenital heart defects, dysmorphic facial features and intellectual developmental disorder (CHDFIDD), is a very rare autosomal dominant genetic condition characterised by congenital heart defects, intellectual disability and characteristic facial features. Those affected typically have motor and language delays, low muscle tone and gastrointestinal dysmotility. Facial features include a wide nasal bridge, widely-spaced eyes, prominent, low-set ears, a flat nose tip and a small mouth. Less common features include congenital spinal abnormalities, hearing loss or seizures.
SYNGAP1-related intellectual disability is a monogenetic developmental and epileptic encephalopathy that affects the central nervous system. Symptoms include intellectual disability, epilepsy, autism, sensory processing deficits, hypotonia and unstable gait.

17q12 microdeletion syndrome, also known as 17q12 deletion syndrome, is a rare chromosomal anomaly caused by the deletion of a small amount of material from a region in the long arm of chromosome 17. It is typified by deletion of the HNF1B gene, resulting in kidney abnormalities and renal cysts and diabetes syndrome. It also has neurocognitive effects, and has been implicated as a genetic factor for autism and schizophrenia.
Autosomal dominant intellectual disability-craniofacial anomalies-cardiac defects syndrome is a rare genetic disorder which is characterized by multi-systemic symptoms primarily affecting the intellect and post-natal development.

Snijders Blok–Campeau syndrome is a genetic disorder caused by mutations in the CHD3 gene. It is characterized by impaired intellectual development, macrocephaly, dysarthria and apraxia of speech, and certain distinctive facial features.

Severe intellectual disability-progressive spastic diplegia syndrome is a rare novel genetic disorder characterized by severe intellectual disabilities, ataxia, craniofacial dysmorphisms, and muscle spasticity. It is a type of autosomal dominant syndromic intellectual disability.
| | This article about a medical condition affecting the nervous system is a stub. You can help Wikipedia by expanding it. |
| | This genetic disorder article is a stub. You can help Wikipedia by expanding it. |