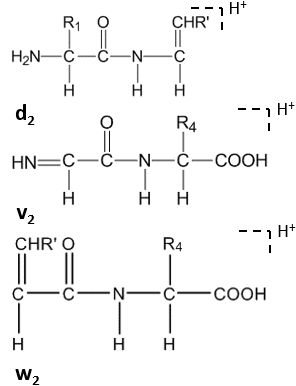
Manual de novo sequencing is labor-intensive and time-consuming. Usually algorithms or programs come with the mass spectrometer instrument are applied for the interpretation of spectra.
Development of de novo sequencing algorithms
An old method is to list all possible peptides for the precursor ion in mass spectrum, and match the mass spectrum for each candidate to the experimental spectrum. The possible peptide that has the most similar spectrum will have the highest chance to be the right sequence. However, the number of possible peptides may be large. For example, a precursor peptide with a molecular weight of 774 has 21,909,046 possible peptides. Even though it is done in the computer, it takes a long time. [17] [18]
Another method is called "subsequencing", which instead of listing whole sequence of possible peptides, matches short sequences of peptides that represent only a part of the complete peptide. When sequences that highly match the fragment ions in the experimental spectrum are found, they are extended by residues one by one to find the best matching. [19] [20] [21] [22]
In the third method, graphical display of the data is applied, in which fragment ions that have the same mass differences of one amino acid residue are connected by lines. In this way, it is easier to get a clear image of ion series of the same type. This method could be helpful for manual de novo peptide sequencing, but doesn't work for high-throughput condition. [23]
The fourth method, which is considered to be successful, is the graph theory. Applying graph theory in de novo peptide sequencing was first mentioned by Bartels. [24] Peaks in the spectrum are transformed into vertices in a graph called "spectrum graph". If two vertices have the same mass difference of one or several amino acids, a directed edge will be applied. The SeqMS algorithm, [25] Lutefisk algorithm, [26] Sherenga algorithm [27] are some examples of this type.
Software packages
As described by Andreotti et al. in 2012, [31] Antilope is a combination of Lagrangian relaxation and an adaptation of Yen's k shortest paths. It is based on 'spectrum graph' method and contains different scoring functions, and can be comparable on the running time and accuracy to "the popular state-of-the-art programs" PepNovo and NovoHMM.
Grossmann et al. [32] presented AUDENS in 2005 as an automated de novo peptide sequencing tool containing a preprocessing module that can recognize signal peaks and noise peaks.
Lutefisk can solve de novo sequencing from CID mass spectra. In this algorithm, significant ions are first found, then determine the N- and C-terminal evidence list. Based on the sequence list, it generates complete sequences in spectra and scores them with the experimental spectrum. However, the result may include several sequence candidates that have only little difference, so it is hard to find the right peptide sequence. A second program, CIDentify, which is a modified version by Alex Taylor of Bill Pearson's FASTA algorithm, can be applied to distinguish those uncertain similar candidates.
Mo et al. presented the MSNovo algorithm in 2007 and proved that it performed "better than existing de novo tools on multiple data sets". [33] This algorithm can do de novo sequencing interpretation of LCQ, LTQ mass spectrometers and of singly, doubly, triply charged ions. Different from other algorithms, it applied a novel scoring function and use a mass array instead of a spectrum graph.
Fisher et al. [34] proposed the NovoHMM method of de novo sequencing. A hidden Markov model (HMM) is applied as a new way to solve de novo sequencing in a Bayesian framework. Instead of scoring for single symbols of the sequence, this method considers posterior probabilities for amino acids. In the paper, this method is proved to have better performance than other popular de novo peptide sequencing methods like PepNovo by a lot of example spectra.
PEAKS is a complete software package for the interpretation of peptide mass spectra. It contains de novo sequencing, database search, PTM identification, homology search and quantification in data analysis. Ma et al. described a new model and algorithm for de novo sequencing in PEAKS, and compared the performance with Lutefisk of several tryptic peptides of standard proteins, by the quadrupole time-of-flight (Q-TOF) mass spectrometer. [35]
PepNovo is a high throughput de novo peptide sequencing tool and uses a probabilistic network as scoring method. It usually takes less than 0.2 seconds for interpretation of one spectrum. Described by Frank et al., PepNovo works better than several popular algorithms like Sherenga, PEAKS, Lutefisk. [36] Now a new version PepNovo+ is available.
Chi et al. presented pNovo+ in 2013 as a new de novo peptide sequencing tool by using complementary HCD and ETD tandem mass spectra. [37] In this method, a component algorithm, pDAG, largely speeds up the acquisition time of peptide sequencing to 0.018s on average, which is three times as fast as the other popular de novo sequencing software.
As described by Jeong et al., compared with other do novo peptide sequencing tools, which works well on only certain types of spectra, UniNovo is a more universal tool that has a good performance on various types of spectra or spectral pairs like CID, ETD, HCD, CID/ETD, etc. It has a better accuracy than PepNovo+ or PEAKS. Moreover, it generates the error rate of the reported peptide sequences. [38]
Ma published Novor in 2015 as a real-time de novo peptide sequencing engine. The tool is sought to improve the de novo speed by an order of magnitude and retain similar accuracy as other de novo tools in the market. On a Macbook Pro laptop, Novor has achieved more than 300 MS/MS spectra per second. [39]
Pevtsov et al. compared the performance of the above five de novo sequencing algorithms: AUDENS, Lutefisk, NovoHMM, PepNovo, and PEAKS . QSTAR and LCQ mass spectrometer data were employed in the analysis, and evaluated by relative sequence distance (RSD) value, which was the similarity between de novo peptide sequencing and true peptide sequence calculated by a dynamic programming method. Results showed that all algorithms had better performance in QSTAR data than on LCQ data, while PEAKS as the best had a success rate of 49.7% in QSTAR data, and NovoHMM as the best had a success rate of 18.3% in LCQ data. The performance order in QSTAR data was PEAKS > Lutefisk, PepNovo > AUDENS, NovoHMM, and in LCQ data was NovoHMM > PepNovo, PEAKS > Lutefisk > AUDENS. Compared in a range of spectrum quality, PEAKS and NovoHMM also showed the best performance in both data among all 5 algorithms. PEAKS and NovoHMM had the best sensitivity in both QSTAR and LCQ data as well. However, no evaluated algorithms exceeded a 50% of exact identification for both data sets. [40]
Recent progress in mass spectrometers made it possible to generate mass spectra of ultra-high resolution . The improved accuracy, together with the increased amount of mass spectrometry data that are being generated, draws the interests of applying deep learning techniques to de novo peptide sequencing. In 2017 Tran et al. proposed DeepNovo, the first deep learning based de novo sequencing software. The benchmark analysis in the original publication demonstrated that DeepNovo outperformed previous methods, including PEAKS, Novor and PepNovo, by a significant margin. DeepNovo is implemented in python with the Tensorflow framework. [41] To represent a mass spectrum as a fixed-dimensional input to the neural-network, DeepNovo discretized each spectrum into a length 150,000 vector. This unnecessarily large spectrum representation, and the single-thread CPU usage in the original implementation, prevents DeepNovo from performing peptide sequencing in real time. To further improve efficiency of de novo peptide sequencing models, Qiao et al. proposed PointNovo in 2020. PointNovo is a python software implemented with the PyTorch framework [42] and it gets rid of the space consuming spectrum-vector-representation adopted by DeepNovo. Comparing with DeepNovo, PointNovo managed to achieve better accuracy and efficiency at the same time by directly representing a spectrum as a set of m/z and intensity pairs.