In organic chemistry, a carboxylic acid is an organic acid that contains a carboxyl group attached to an R-group. The general formula of a carboxylic acid is often written as R−COOH or R−CO2H, sometimes as R−C(O)OH with R referring to the alkyl, alkenyl, aryl, or other group. Carboxylic acids occur widely. Important examples include the amino acids and fatty acids. Deprotonation of a carboxylic acid gives a carboxylate anion.
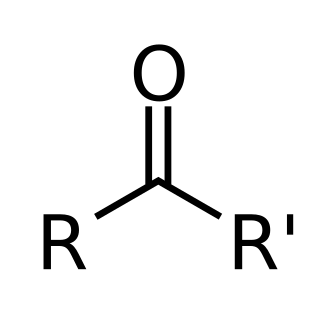
In organic chemistry, a ketone is an organic compound with the structure R−C(=O)−R', where R and R' can be a variety of carbon-containing substituents. Ketones contain a carbonyl group −C(=O)−. The simplest ketone is acetone, with the formula (CH3)2CO. Many ketones are of great importance in biology and in industry. Examples include many sugars (ketoses), many steroids, and the solvent acetone.

In organic chemistry, an aldehyde is an organic compound containing a functional group with the structure R−CH=O. The functional group itself can be referred to as an aldehyde but can also be classified as a formyl group. Aldehydes are a common motif in many chemicals important in technology and biology.

In organic chemistry, a dicarbonyl is a molecule containing two carbonyl groups. Although this term could refer to any organic compound containing two carbonyl groups, it is used more specifically to describe molecules in which both carbonyls are in close enough proximity that their reactivity is changed, such as 1,2-, 1,3-, and 1,4-dicarbonyls. Their properties often differ from those of monocarbonyls, and so they are usually considered functional groups of their own. These compounds can have symmetrical or unsymmetrical substituents on each carbonyl, and may also be functionally symmetrical or unsymmetrical.

The aldol reaction is a reaction in organic chemistry that combines two carbonyl compounds to form a new β-hydroxy carbonyl compound. Its simplest form might involve the nucleophilic addition of an enolized ketone to another:

An enamine is an unsaturated compound derived by the condensation of an aldehyde or ketone with a secondary amine. Enamines are versatile intermediates.

In organometallic chemistry, organolithium reagents are chemical compounds that contain carbon–lithium (C–Li) bonds. These reagents are important in organic synthesis, and are frequently used to transfer the organic group or the lithium atom to the substrates in synthetic steps, through nucleophilic addition or simple deprotonation. Organolithium reagents are used in industry as an initiator for anionic polymerization, which leads to the production of various elastomers. They have also been applied in asymmetric synthesis in the pharmaceutical industry. Due to the large difference in electronegativity between the carbon atom and the lithium atom, the C−Li bond is highly ionic. Owing to the polar nature of the C−Li bond, organolithium reagents are good nucleophiles and strong bases. For laboratory organic synthesis, many organolithium reagents are commercially available in solution form. These reagents are highly reactive, and are sometimes pyrophoric.

In organic chemistry, the Michael reaction or Michael 1,4 addition is a reaction between a Michael donor and a Michael acceptor to produce a Michael adduct by creating a carbon-carbon bond at the acceptor's β-carbon. It belongs to the larger class of conjugate additions and is widely used for the mild formation of carbon-carbon bonds.

In organic chemistry, enolates are organic anions derived from the deprotonation of carbonyl compounds. Rarely isolated, they are widely used as reagents in the synthesis of organic compounds.
The Claisen condensation is a carbon–carbon bond forming reaction that occurs between two esters or one ester and another carbonyl compound in the presence of a strong base. The reaction produces a β-keto ester or a β-diketone. It is named after Rainer Ludwig Claisen, who first published his work on the reaction in 1887. The reaction has often been displaced by diketene-based chemistry, which affords acetoacetic esters.
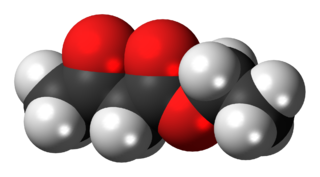
The organic compound ethyl acetoacetate (EAA) is the ethyl ester of acetoacetic acid. It is a colorless liquid. It is widely used as a chemical intermediate in the production of a wide variety of compounds. It is used as a flavoring for food.

Nucleophilic conjugate addition is a type of organic reaction. Ordinary nucleophilic additions or 1,2-nucleophilic additions deal mostly with additions to carbonyl compounds. Simple alkene compounds do not show 1,2 reactivity due to lack of polarity, unless the alkene is activated with special substituents. With α,β-unsaturated carbonyl compounds such as cyclohexenone it can be deduced from resonance structures that the β position is an electrophilic site which can react with a nucleophile. The negative charge in these structures is stored as an alkoxide anion. Such a nucleophilic addition is called a nucleophilic conjugate addition or 1,4-nucleophilic addition. The most important active alkenes are the aforementioned conjugated carbonyls and acrylonitriles.
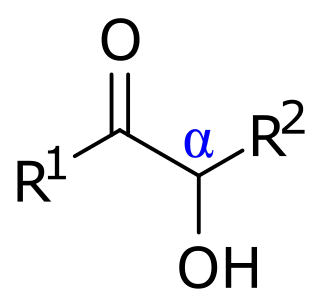
In organic chemistry, acyloins or α-hydroxy ketones are a class of organic compounds of the general form R−C(=O)−CR'(OH)−R", composed of a hydroxy group adjacent to a ketone group. The name acyloin is derived from the fact that they are formally derived from reductive coupling of carboxylic acyl groups. They are one of the two main classes of hydroxy ketones, distinguished by the position of the hydroxy group relative to the ketone; in this form, the hydroxy is on the alpha carbon, explaining the secondary name of α-hydroxy ketone.

The Dakin oxidation (or Dakin reaction) is an organic redox reaction in which an ortho- or para-hydroxylated phenyl aldehyde (2-hydroxybenzaldehyde or 4-hydroxybenzaldehyde) or ketone reacts with hydrogen peroxide (H2O2) in base to form a benzenediol and a carboxylate. Overall, the carbonyl group is oxidised, whereas the H2O2 is reduced.
In organic chemistry, aldol reactions are acid- or base-catalyzed reactions of aldehydes or ketones.

In organic chemistry, vinylogy is the transmission of electronic effects through a conjugated organic bonding system. The concept was introduced in 1926 by Ludwig Claisen to explain the acidic properties of formylacetone and related ketoaldehydes. Formylacetone, technically CH3(C=O)CH2CH=O, only exists in the ionized form CH3(C−O−)=CH−CH=O or CH3(C=O)−CH=CH−O−. Its adjectival form, vinylogous, is used to describe functional groups in which the standard moieties of the group are separated by a carbon–carbon double bond.
Selenoxide elimination is a method for the chemical synthesis of alkenes from selenoxides. It is most commonly used to synthesize α,β-unsaturated carbonyl compounds from the corresponding saturated analogues. It is mechanistically related to the Cope reaction.

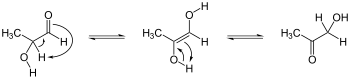
Alpha-substitution reactions occur at the position next to the carbonyl group, the α-position, and involve the substitution of an α hydrogen atom by an electrophile, E, through either an enol or enolate ion intermediate.
In organic chemistry, the Conia-ene reaction is an intramolecular cyclization reaction between an enolizable carbonyl such as an ester or ketone and an alkyne or alkene, giving a cyclic product with a new carbon-carbon bond. As initially reported by J. M. Conia and P. Le Perchec, the Conia-ene reaction is a heteroatom analog of the ene reaction that uses an enol as the ene component. Like other pericyclic reactions, the original Conia-ene reaction required high temperatures to proceed, limiting its wider application. However, subsequent improvements, particularly in metal catalysis, have led to significant expansion of reaction scope. Consequently, various forms of the Conia-ene reaction have been employed in the synthesis of complex molecules and natural products.
The ketimine Mannich reaction is an asymmetric synthetic technique using differences in starting material to push a Mannich reaction to create an enantiomeric product with steric and electronic effects, through the creation of a ketimine group. Typically, this is done with a reaction with proline or another nitrogen-containing heterocycle, which control chirality with that of the catalyst. This has been theorized to be caused by the restriction of undesired (E)-isomer by preventing the ketone from accessing non-reactive tautomers. Generally, a Mannich reaction is the combination of an amine, a ketone with a β-acidic proton and aldehyde to create a condensed product in a β-addition to the ketone. This occurs through an attack on the ketone with a suitable catalytic-amine unto its electron-starved carbon, from which an imine is created. This then undergoes electrophilic addition with a compound containing an acidic proton. It is theoretically possible for either of the carbonyl-containing molecules to create diastereomers, but with the addition of catalysts which restrict addition as of the enamine creation, it is possible to extract a single product with limited purification steps and in some cases as reported by List et al.; practical one-pot syntheses are possible. The process of selecting a carbonyl-group gives the reaction a direct versus indirect distinction, wherein the latter case represents pre-formed products restricting the reaction's pathway and the other does not. Ketimines selects a reaction group, and circumvent a requirement for indirect pathways.