
The United States Food and Drug Administration is a federal agency of the Department of Health and Human Services. The FDA is responsible for protecting and promoting public health through the control and supervision of food safety, tobacco products, dietary supplements, prescription and over-the-counter pharmaceutical drugs (medications), vaccines, biopharmaceuticals, blood transfusions, medical devices, electromagnetic radiation emitting devices (ERED), cosmetics, animal foods & feed and veterinary products.

The United States Federal Food, Drug, and Cosmetic Act is a set of laws passed by the United States Congress in 1938 giving authority to the U.S. Food and Drug Administration (FDA) to oversee the safety of food, drugs, medical devices, and cosmetics. A principal author of this law was Royal S. Copeland, a three-term U.S. senator from New York. In 1968, the Electronic Product Radiation Control provisions were added to the FD&C. Also in that year the FDA formed the Drug Efficacy Study Implementation (DESI) to incorporate into FD&C regulations the recommendations from a National Academy of Sciences investigation of effectiveness of previously marketed drugs. The act has been amended many times, most recently to add requirements about bioterrorism preparations.

A medical device is any device intended to be used for medical purposes. Significant potential for hazards are inherent when using a device for medical purposes and thus medical devices must be proved safe and effective with reasonable assurance before regulating governments allow marketing of the device in their country. As a general rule, as the associated risk of the device increases the amount of testing required to establish safety and efficacy also increases. Further, as associated risk increases the potential benefit to the patient must also increase.
An investigational device exemption (IDE) allows an investigational device to be used in order to collect safety and effectiveness data required to support a premarket approval (PMA) application or a premarket notification [510(k)] submission to Food and Drug Administration (FDA). Clinical studies are most often conducted to support a PMA. Only a small percentage of 510(k)'s require clinical data to support the application. Investigational use also includes clinical evaluation of certain modifications or new intended uses of legally marketed devices. All clinical evaluations of investigational devices, unless exempt, must have an approved IDE before the study is initiated.
A drug recall removes a prescription or over-the-counter drug from the market. Drug recalls in the United States are made by the FDA or the creators of the drug when certain criteria are met. When a drug recall is made, the drug is removed from the market and potential legal action can be taken depending on the severity of the drug recall.
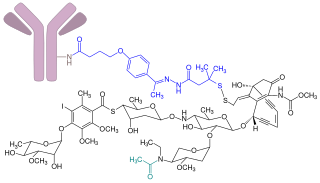
Gemtuzumab ozogamicin, sold under the brand name Mylotarg, is an antibody-drug conjugate that is used to treat acute myeloid leukemia.

The Center for Veterinary Medicine (CVM) is a branch of the U.S. Food and Drug Administration (FDA) that regulates the manufacture and distribution of food, food additives, and drugs that will be given to animals. These include animals from which human foods are derived, as well as food additives and drugs for pets or companion animals. CVM is responsible for regulating drugs, devices, and food additives given to, or used on, over one hundred million companion animals, plus millions of poultry, cattle, swine, and minor animal species. Minor animal species include animals other than cattle, swine, chickens, turkeys, horses, dogs, and cats.
Clinical research is a branch of healthcare science that determines the safety and effectiveness (efficacy) of medications, devices, diagnostic products and treatment regimens intended for human use. These may be used for prevention, treatment, diagnosis or for relieving symptoms of a disease. Clinical research is different from clinical practice. In clinical practice established treatments are used, while in clinical research evidence is collected to establish a treatment.
Postmarketing surveillance (PMS), also known as post market surveillance, is the practice of monitoring the safety of a pharmaceutical drug or medical device after it has been released on the market and is an important part of the science of pharmacovigilance. Since drugs and medical devices are approved on the basis of clinical trials, which involve relatively small numbers of people who have been selected for this purpose – meaning that they normally do not have other medical conditions which may exist in the general population – postmarketing surveillance can further refine, or confirm or deny, the safety of a drug or device after it is used in the general population by large numbers of people who have a wide variety of medical conditions.

The Center for Devices and Radiological Health (CDRH) is the branch of the United States Food and Drug Administration (FDA) responsible for the premarket approval of all medical devices, as well as overseeing the manufacturing, performance and safety of these devices. The CDRH also oversees the radiation safety performance of non-medical devices which emit certain types of electromagnetic radiation, such as cellular phones and microwave ovens.
Expanded access or compassionate use is the use of an unapproved drug or medical device under special forms of investigational new drug applications (IND) or IDE application for devices, outside of a clinical trial, by people with serious or life-threatening conditions who do not meet the enrollment criteria for the clinical trial in progress.
President of the United States George W. Bush signed the Food and Drug Administration Amendments Act of 2007 (FDAAA) on September 27, 2007. This law reviewed, expanded, and reaffirmed several existing pieces of legislation regulating the FDA. These changes allow the FDA to perform more comprehensive reviews of potential new drugs and devices. It was sponsored by Reps. Joe Barton and Frank Pallone and passed unanimously by the Senate.
Priority review is a program of the United States Food and Drug Administration (FDA) to expedite the review process for drugs that are expected to have a particularly great impact on the treatment of a disease. The priority review voucher program is a program that grants a voucher for priority review to a drug developer as an incentive to develop treatments for disease indications with limited profitability.
The following outline is provided as an overview of and topical guide to clinical research:
The United States Food and Drug Administration Modernization Act of 1997 (FDAMA) amended the Federal Food, Drug, and Cosmetic Act. This act is related to the regulation of food, drugs, devices, and biological products by the FDA. These changes were made in order to recognize the changes in the way the FDA would be operating in the 21st century. The main focus of this is the acknowledgment in the advancement of technological, trade, and public health complexities.
Avita Medical is a clinical and commercial company developing and marketing a range of respiratory and regenerative products. The first regenerative medicine product brought to the market by Avita Medical was ReCell spray-on skin for the treatment of burns. The two latest products are ReNovaCell, for Aesthetics and Plastic applications including skin trauma, and ReGenerCell for the treatment of chronic wounds. The Avita Medical regenerative product range is currently marketed in Europe, the Middle East, Africa (EMEA) and Australia.
This article is about the history of the United States Food and Drug Administration.
The Food and Drug Administration Safety and Innovation Act of 2012 (FDASIA) is a piece of American regulatory legislation signed into law on July 9, 2012. It gives the United States Food and Drug Administration (FDA) the authority to collect user fees from the medical industry to fund reviews of innovator drugs, medical devices, generic drugs and biosimilar biologics. It also creates the breakthrough therapy designation program and extends the priority review voucher program to make eligible rare pediatric diseases. The measure was passed by 96 senators voting for and one voting against.
Due to the many regulations in the industry, the design of medical devices presents significant challenges from both engineering and legal perspectives.
In February 2021, the U.S. Food and Drug Administration (FDA) approved the Patient Specific Talus Spacer 3D-printed talus implant for humanitarian use. The Patient Specific Talus Spacer is the first in the world and first-of-its-kind implant to replace the talus—the bone in the ankle joint that connects the leg and the foot—for the treatment of avascular necrosis (AVN) of the ankle joint, a serious and progressive condition that causes the death of bone tissue stemming from a lack of blood supply to the area. The implant provides a joint-sparing alternative to other surgical interventions commonly used in late-stage AVN that may disable motion of the ankle joint.