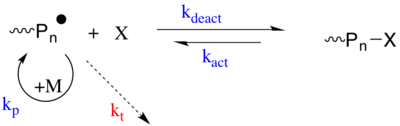In polymer chemistry, emulsion polymerization is a type of radical polymerization that usually starts with an emulsion incorporating water, monomers, and surfactants. The most common type of emulsion polymerization is an oil-in-water emulsion, in which droplets of monomer are emulsified in a continuous phase of water. Water-soluble polymers, such as certain polyvinyl alcohols or hydroxyethyl celluloses, can also be used to act as emulsifiers/stabilizers. The name "emulsion polymerization" is a misnomer that arises from a historical misconception. Rather than occurring in emulsion droplets, polymerization takes place in the latex/colloid particles that form spontaneously in the first few minutes of the process. These latex particles are typically 100 nm in size, and are made of many individual polymer chains. The particles are prevented from coagulating with each other because each particle is surrounded by the surfactant ('soap'); the charge on the surfactant repels other particles electrostatically. When water-soluble polymers are used as stabilizers instead of soap, the repulsion between particles arises because these water-soluble polymers form a 'hairy layer' around a particle that repels other particles, because pushing particles together would involve compressing these chains.

In polymer chemistry, a copolymer is a polymer derived from more than one species of monomer. The polymerization of monomers into copolymers is called copolymerization. Copolymers obtained from the copolymerization of two monomer species are sometimes called bipolymers. Those obtained from three and four monomers are called terpolymers and quaterpolymers, respectively. Copolymers can be characterized by a variety of techniques such as NMR spectroscopy and size-exclusion chromatography to determine the molecular size, weight, properties, and composition of the material.
Chain-growth polymerization (AE) or chain-growth polymerisation (BE) is a polymerization technique where unsaturated monomer molecules add onto the active site on a growing polymer chain one at a time. There are a limited number of these active sites at any moment during the polymerization which gives this method its key characteristics.
In polymer chemistry, free-radical polymerization (FRP) is a method of polymerization by which a polymer forms by the successive addition of free-radical building blocks. Free radicals can be formed by a number of different mechanisms, usually involving separate initiator molecules. Following its generation, the initiating free radical adds (nonradical) monomer units, thereby growing the polymer chain.
In polymer chemistry, anionic addition polymerization is a form of chain-growth polymerization or addition polymerization that involves the polymerization of monomers initiated with anions. The type of reaction has many manifestations, but traditionally vinyl monomers are used. Often anionic polymerization involves living polymerizations, which allows control of structure and composition.
Atom transfer radical polymerization (ATRP) is an example of a reversible-deactivation radical polymerization. Like its counterpart, ATRA, or atom transfer radical addition, ATRP is a means of forming a carbon-carbon bond with a transition metal catalyst. Polymerization from this method is called atom transfer radical addition polymerization (ATRAP). As the name implies, the atom transfer step is crucial in the reaction responsible for uniform polymer chain growth. ATRP was independently discovered by Mitsuo Sawamoto and by Krzysztof Matyjaszewski and Jin-Shan Wang in 1995.

Reversible addition−fragmentation chain-transfer or RAFT polymerization is one of several kinds of reversible-deactivation radical polymerization. It makes use of a chain-transfer agent (CTA) in the form of a thiocarbonylthio compound to afford control over the generated molecular weight and polydispersity during a free-radical polymerization. Discovered at the Commonwealth Scientific and Industrial Research Organisation (CSIRO) of Australia in 1998, RAFT polymerization is one of several living or controlled radical polymerization techniques, others being atom transfer radical polymerization (ATRP) and nitroxide-mediated polymerization (NMP), etc. RAFT polymerization uses thiocarbonylthio compounds, such as dithioesters, thiocarbamates, and xanthates, to mediate the polymerization via a reversible chain-transfer process. As with other controlled radical polymerization techniques, RAFT polymerizations can be performed under conditions that favor low dispersity and a pre-chosen molecular weight. RAFT polymerization can be used to design polymers of complex architectures, such as linear block copolymers, comb-like, star, brush polymers, dendrimers and cross-linked networks.
Solution polymerization is a method of industrial polymerization. In this procedure, a monomer is dissolved in a non-reactive solvent that contains a catalyst or initiator.
Catalytic chain transfer (CCT) is a process that can be incorporated into radical polymerization to obtain greater control over the resulting products.
Cobalt based catalysts, when used in radical polymerization, have several main advantages especially in slowing down the reaction rate, allowing for the synthesis of polymers with peculiar properties. As starting the reaction does need a real radical initiator, the cobalt species is not the only used catalyst, it is a mediator. For this reason this type of reaction is referred to as cobalt mediated radical polymerization.
Living cationic polymerization is a living polymerization technique involving cationic propagating species. It enables the synthesis of very well defined polymers and of polymers with unusual architecture such as star polymers and block copolymers and living cationic polymerization is therefore as such of commercial and academic interest.
Living free radical polymerization is a type of living polymerization where the active polymer chain end is a free radical. Several methods exist. IUPAC recommends to use the term "reversible-deactivation radical polymerization" instead of "living free radical polymerization", though the two terms are not synonymous.
In polymer chemistry, cationic polymerization is a type of chain growth polymerization in which a cationic initiator transfers charge to a monomer, which then becomes reactive. This reactive monomer goes on to react similarly with other monomers to form a polymer. The types of monomers necessary for cationic polymerization are limited to alkenes with electron-donating substituents and heterocycles. Similar to anionic polymerization reactions, cationic polymerization reactions are very sensitive to the type of solvent used. Specifically, the ability of a solvent to form free ions will dictate the reactivity of the propagating cationic chain. Cationic polymerization is used in the production of polyisobutylene and poly(N-vinylcarbazole) (PVK).
In polymer chemistry, chain walking (CW) or chain running or chain migration is a mechanism that operates during some alkene polymerization reactions. CW can be also considered as a specific case of intermolecular chain transfer. This reaction gives rise to branched and hyperbranched/dendritic hydrocarbon polymers. This process is also characterized by accurate control of polymer architecture and topology. The extent of CW, displayed in the number of branches formed and positions of branches on the polymers are controlled by the choice of a catalyst. The potential applications of polymers formed by this reaction are diverse, from drug delivery to phase transfer agents, nanomaterials, and catalysis.
In polymer chemistry, reversible-deactivation radical polymerizations (RDRPs) are members of the class of reversible-deactivation polymerizations which exhibit much of the character of living polymerizations, but cannot be categorized as such as they are not without chain transfer or chain termination reactions. Several different names have been used in literature, which are:
In polymer chemistry, ionic polymerization is a chain-growth polymerization in which active centers are ions or ion pairs. It can be considered as an alternative to radical polymerization, and may refer to anionic polymerization or cationic polymerization.

A sequence-controlled polymer is a macromolecule, in which the sequence of monomers is controlled to some degree. This control can be absolute but not necessarily. In other words, a sequence-controlled polymer can be uniform or non-uniform (Ð>1). For example, an alternating copolymer synthesized by radical polymerization is a sequence-controlled polymer, even if it is also a non-uniform polymer, in which chains have different chain-lengths and slightly different compositions. A biopolymer with a perfectly-defined primary structure is also a sequence-controlled polymer. However, in the case of uniform macromolecules, the term sequence-defined polymer can also be used.

In polymer chemistry, graft polymers are segmented copolymers with a linear backbone of one composite and randomly distributed branches of another composite. The picture labeled "graft polymer" shows how grafted chains of species B are covalently bonded to polymer species A. Although the side chains are structurally distinct from the main chain, the individual grafted chains may be homopolymers or copolymers. Graft polymers have been synthesized for many decades and are especially used as impact resistant materials, thermoplastic elastomers, compatibilizers, or emulsifiers for the preparation of stable blends or alloys. One of the better-known examples of a graft polymer is a component used in high impact polystyrene, consisting of a polystyrene backbone with polybutadiene grafted chains.
Functionalized polyolefins are olefin polymers with polar and nonpolar functionalities attached onto the polymer backbone. There has been an increased interest in functionalizing polyolefins due to their increased usage in everyday life. Polyolefins are virtually ubiquitous in everyday life, from consumer food packaging to biomedical applications; therefore, efforts must be made to study catalytic pathways towards the attachment of various functional groups onto polyolefins in order to affect the material's physical properties.
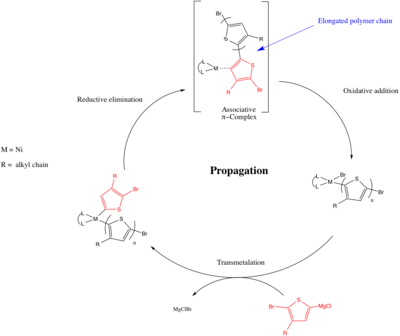
Catalyst transfer polymerization (CTP), or catalyst transfer polycondensation, is a type of living chain-growth polymerization that is used for synthesizing conjugated polymers. Benefits to using CTP over other methods are low polydispersity and control over number average molecular weight in the resulting polymers. Very few monomers have been demonstrated to undergo CTP.