Related Research Articles
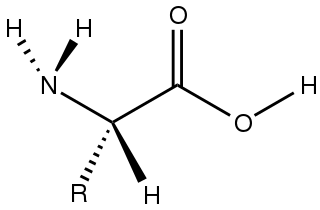
Amino acids are organic compounds that contain both amino and carboxylic acid functional groups. Although over 500 amino acids exist in nature, by far the most important are the 22 α-amino acids incorporated into proteins. Only these 22 appear in the genetic code of life.

The genetic code is the set of rules used by living cells to translate information encoded within genetic material into proteins. Translation is accomplished by the ribosome, which links proteinogenic amino acids in an order specified by messenger RNA (mRNA), using transfer RNA (tRNA) molecules to carry amino acids and to read the mRNA three nucleotides at a time. The genetic code is highly similar among all organisms and can be expressed in a simple table with 64 entries.
Non-coding DNA (ncDNA) sequences are components of an organism's DNA that do not encode protein sequences. Some non-coding DNA is transcribed into functional non-coding RNA molecules. Other functional regions of the non-coding DNA fraction include regulatory sequences that control gene expression; scaffold attachment regions; origins of DNA replication; centromeres; and telomeres. Some non-coding regions appear to be mostly nonfunctional, such as introns, pseudogenes, intergenic DNA, and fragments of transposons and viruses. Regions that are completely nonfunctional are called junk DNA.
Molecular evolution is the process of change in the sequence composition of cellular molecules such as DNA, RNA, and proteins across generations. The field of molecular evolution uses principles of evolutionary biology and population genetics to explain patterns in these changes. Major topics in molecular evolution concern the rates and impacts of single nucleotide changes, neutral evolution vs. natural selection, origins of new genes, the genetic nature of complex traits, the genetic basis of speciation, the evolution of development, and ways that evolutionary forces influence genomic and phenotypic changes.

In biology, translation is the process in living cells in which proteins are produced using RNA molecules as templates. The generated protein is a sequence of amino acids. This sequence is determined by the sequence of nucleotides in the RNA. The nucleotides are considered three at a time. Each such triple results in addition of one specific amino acid to the protein being generated. The matching from nucleotide triple to amino acid is called the genetic code. The translation is performed by a large complex of functional RNA and proteins called ribosomes. The entire process is called gene expression.

Proteinogenic amino acids are amino acids that are incorporated biosynthetically into proteins during translation. The word "proteinogenic" means "protein creating". Throughout known life, there are 22 genetically encoded (proteinogenic) amino acids, 20 in the standard genetic code and an additional 2 that can be incorporated by special translation mechanisms.

In genetics and bioinformatics, a single-nucleotide polymorphism is a germline substitution of a single nucleotide at a specific position in the genome that is present in a sufficiently large fraction of considered population.

A point mutation is a genetic mutation where a single nucleotide base is changed, inserted or deleted from a DNA or RNA sequence of an organism's genome. Point mutations have a variety of effects on the downstream protein product—consequences that are moderately predictable based upon the specifics of the mutation. These consequences can range from no effect to deleterious effects, with regard to protein production, composition, and function.

A leucine zipper is a common three-dimensional structural motif in proteins. They were first described by Landschulz and collaborators in 1988 when they found that an enhancer binding protein had a very characteristic 30-amino acid segment and the display of these amino acid sequences on an idealized alpha helix revealed a periodic repetition of leucine residues at every seventh position over a distance covering eight helical turns. The polypeptide segments containing these periodic arrays of leucine residues were proposed to exist in an alpha-helical conformation and the leucine side chains from one alpha helix interdigitate with those from the alpha helix of a second polypeptide, facilitating dimerization.

In evolutionary biology, conserved sequences are identical or similar sequences in nucleic acids or proteins across species, or within a genome, or between donor and receptor taxa. Conservation indicates that a sequence has been maintained by natural selection.
A DNA-binding domain (DBD) is an independently folded protein domain that contains at least one structural motif that recognizes double- or single-stranded DNA. A DBD can recognize a specific DNA sequence or have a general affinity to DNA. Some DNA-binding domains may also include nucleic acids in their folded structure.
In biology, the word gene has two meanings. The Mendelian gene is a basic unit of heredity. The molecular gene is a sequence of nucleotides in DNA, that is transcribed to produce a functional RNA. There are two types of molecular genes: protein-coding genes and non-coding genes.
Gene structure is the organisation of specialised sequence elements within a gene. Genes contain most of the information necessary for living cells to survive and reproduce. In most organisms, genes are made of DNA, where the particular DNA sequence determines the function of the gene. A gene is transcribed (copied) from DNA into RNA, which can either be non-coding (ncRNA) with a direct function, or an intermediate messenger (mRNA) that is then translated into protein. Each of these steps is controlled by specific sequence elements, or regions, within the gene. Every gene, therefore, requires multiple sequence elements to be functional. This includes the sequence that actually encodes the functional protein or ncRNA, as well as multiple regulatory sequence regions. These regions may be as short as a few base pairs, up to many thousands of base pairs long.

Directed evolution (DE) is a method used in protein engineering that mimics the process of natural selection to steer proteins or nucleic acids toward a user-defined goal. It consists of subjecting a gene to iterative rounds of mutagenesis, selection and amplification. It can be performed in vivo, or in vitro. Directed evolution is used both for protein engineering as an alternative to rationally designing modified proteins, as well as for experimental evolution studies of fundamental evolutionary principles in a controlled, laboratory environment.

In molecular biology and genetics, DNA annotation or genome annotation is the process of describing the structure and function of the components of a genome, by analyzing and interpreting them in order to extract their biological significance and understand the biological processes in which they participate. Among other things, it identifies the locations of genes and all the coding regions in a genome and determines what those genes do.
In molecular biology, the cold-shock domain (CSD) is a protein domain of about 70 amino acids which has been found in prokaryotic and eukaryotic DNA-binding proteins. Part of this domain is highly similar to the RNP-1 RNA-binding motif.

Edward Nikolayevich Trifonov is a Russian-born Israeli molecular biophysicist and a founder of Israeli bioinformatics. In his research, he specializes in the recognition of weak signal patterns in biological sequences and is known for his unorthodox scientific methods.
Proline-rich protein 21 (PRR21) is a protein of the family of proline-rich proteins. It is encoded by the PRR21 gene, which is found on human chromosome 2, band 2q37.3. The gene exists in several species, both vertebrates and invertebrates, including humans. However, the protein have few conserved regions among species.

An array of protein tandem repeats is defined as several adjacent copies having the same or similar sequence motifs. These periodic sequences are generated by internal duplications in both coding and non-coding genomic sequences. Repetitive units of protein tandem repeats are considerably diverse, ranging from the repetition of a single amino acid to domains of 100 or more residues.
References
- 1 2 Wootton JC (September 1994). "Non-globular domains in protein sequences: Automated segmentation using complexity measures". Computers & Chemistry. 18 (3): 269–285. doi:10.1016/0097-8485(94)85023-2. PMID 7952898.
- 1 2 Mier P, Paladin L, Tamana S, Petrosian S, Hajdu-Soltész B, Urbanek A, Gruca A, Plewczynski D, Grynberg M, Bernadó P, Gáspári Z, Ouzounis CA, Promponas VJ, Kajava AV, Hancock JM, Tosatto SC, Dosztanyi Z, Andrade-Navarro MA (30 January 2019). "Disentangling the complexity of low complexity proteins". Brief Bioinform. 21 (2): 458–472. doi: 10.1093/bib/bbz007 . PMC 7299295 . PMID 30698641.
- 1 2 Huntley MA, Golding GB (2002-07-01). "Simple sequences are rare in the Protein Data Bank". Proteins: Structure, Function, and Genetics. 48 (1): 134–140. doi:10.1002/prot.10150. ISSN 0887-3585. PMID 12012345. S2CID 42193081.
- ↑ Kumari B, Kumar R, Kumar M (2015). "Low complexity and disordered regions of proteins have different structural and amino acid preferences". Molecular BioSystems. 11 (2): 585–594. doi:10.1039/C4MB00425F. ISSN 1742-206X. PMID 25468592.
- ↑ Luo H, Nijveen H (2014-07-01). "Understanding and identifying amino acid repeats". Briefings in Bioinformatics. 15 (4): 582–591. doi:10.1093/bib/bbt003. ISSN 1467-5463. PMC 4103538 . PMID 23418055.
- ↑ Matsushima N, Yoshida H, Kumaki Y, Kamiya M, Tanaka T, Kretsinger YI (2008-11-30). "Flexible Structures and Ligand Interactions of Tandem Repeats Consisting of Proline, Glycine, Asparagine, Serine, and/or Threonine Rich Oligopeptides in Proteins". Current Protein & Peptide Science. 9 (6): 591–610. doi:10.2174/138920308786733886. PMID 19075749 . Retrieved 2020-11-03.
- ↑ Adzhubei AA, Sternberg MJ, Makarov AA (June 2013). "Polyproline-II Helix in Proteins: Structure and Function". Journal of Molecular Biology. 425 (12): 2100–2132. doi:10.1016/j.jmb.2013.03.018. PMID 23507311.
- 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 Ntountoumi C, Vlastaridis P, Mossialos D, Stathopoulos C, Iliopoulos I, Promponas V, Oliver SG, Amoutzias GD (2019-11-04). "Low complexity regions in the proteins of prokaryotes perform important functional roles and are highly conserved". Nucleic Acids Research. 47 (19): 9998–10009. doi:10.1093/nar/gkz730. ISSN 0305-1048. PMC 6821194 . PMID 31504783.
 Text was copied from this source, which is available under a Creative Commons Attribution 4.0 International License.
Text was copied from this source, which is available under a Creative Commons Attribution 4.0 International License. - ↑ Karlin S, Brocchieri L, Bergman A, Mrazek J, Gentles AJ (2002-01-08). "Amino acid runs in eukaryotic proteomes and disease associations". Proceedings of the National Academy of Sciences. 99 (1): 333–338. Bibcode:2002PNAS...99..333K. doi: 10.1073/pnas.012608599 . ISSN 0027-8424. PMC 117561 . PMID 11782551.
- ↑ Mirkin SM (2007-06-21). "Expandable DNA repeats and human disease". Nature. 447 (7147): 932–940. Bibcode:2007Natur.447..932M. doi:10.1038/nature05977. ISSN 0028-0836. PMID 17581576. S2CID 4397592.
- ↑ Kumari B, Kumar R, Chauhan V, Kumar M (2018-10-30). "Comparative functional analysis of proteins containing low-complexity predicted amyloid regions". PeerJ. 6: e5823. doi: 10.7717/peerj.5823 . ISSN 2167-8359. PMC 6214233 . PMID 30397544.
- ↑ So CR, Fears KP, Leary DH, Scancella JM, Wang Z, Liu JL, Orihuela B, Rittschof D, Spillmann CM, Wahl KJ (2016-11-08). "Sequence basis of Barnacle Cement Nanostructure is Defined by Proteins with Silk Homology". Scientific Reports. 6 (1): 36219. Bibcode:2016NatSR...636219S. doi:10.1038/srep36219. ISSN 2045-2322. PMC 5099703 . PMID 27824121.
- ↑ Haritos VS, Niranjane A, Weisman S, Trueman HE, Sriskantha A, Sutherland TD (2010-11-07). "Harnessing disorder: onychophorans use highly unstructured proteins, not silks, for prey capture". Proceedings of the Royal Society B: Biological Sciences. 277 (1698): 3255–3263. doi:10.1098/rspb.2010.0604. ISSN 0962-8452. PMC 2981920 . PMID 20519222.
- ↑ Brewer S, Tolley M, Trayer I, Barr G, Dorman C, Hannavy K, Higgins C, Evans J, Levine B, Wormald M (1990-12-20). "Structure and function of X-Pro dipeptide repeats in the TonB proteins of Salmonella typhimurium and Escherichia coli". Journal of Molecular Biology. 216 (4): 883–895. doi:10.1016/S0022-2836(99)80008-4. PMID 2266560.
- ↑ Robison AD, Sun S, Poyton MF, Johnson GA, Pellois J, Jungwirth P, Vazdar M, Cremer PS (2016-09-08). "Polyarginine Interacts More Strongly and Cooperatively than Polylysine with Phospholipid Bilayers". The Journal of Physical Chemistry B. 120 (35): 9287–9296. doi:10.1021/acs.jpcb.6b05604. ISSN 1520-6106. PMC 5912336 . PMID 27571288.
- 1 2 Zhu ZY, Karlin S (1996-08-06). "Clusters of charged residues in protein three-dimensional structures". Proceedings of the National Academy of Sciences. 93 (16): 8350–8355. Bibcode:1996PNAS...93.8350Z. doi: 10.1073/pnas.93.16.8350 . ISSN 0027-8424. PMC 38674 . PMID 8710874.
- ↑ Kushwaha AK, Grove A (2013-02-01). "C-terminal low-complexity sequence repeats of Mycobacterium smegmatis Ku modulate DNA binding". Bioscience Reports. 33 (1): 175–84. doi:10.1042/BSR20120105. ISSN 0144-8463. PMC 3553676 . PMID 23167261.
- ↑ Frugier M, Bour T, Ayach M, Santos MA, Rudinger-Thirion J, Théobald-Dietrich A, Pizzi E (2010-01-21). "Low Complexity Regions behave as tRNA sponges to help co-translational folding of plasmodial proteins". FEBS Letters. 584 (2): 448–454. doi: 10.1016/j.febslet.2009.11.004 . PMID 19900443. S2CID 24172658.
- ↑ Tyedmers J, Mogk A, Bukau B (November 2010). "Cellular strategies for controlling protein aggregation". Nature Reviews Molecular Cell Biology. 11 (11): 777–788. doi:10.1038/nrm2993. ISSN 1471-0072. PMID 20944667. S2CID 22449895.
- ↑ Ling J, Cho C, Guo L, Aerni HR, Rinehart J, Söll D (2012-12-14). "Protein Aggregation Caused by Aminoglycoside Action Is Prevented by a Hydrogen Peroxide Scavenger". Molecular Cell. 48 (5): 713–722. doi:10.1016/j.molcel.2012.10.001. PMC 3525788 . PMID 23122414.
- 1 2 Haerty W, Golding GB (October 2010). Bonen L (ed.). "Low-complexity sequences and single amino acid repeats: not just "junk" peptide sequences". Genome. 53 (10): 753–762. doi:10.1139/G10-063. ISSN 0831-2796. PMID 20962881.
- ↑ Faux NG (2005-03-21). "Functional insights from the distribution and role of homopeptide repeat-containing proteins". Genome Research. 15 (4): 537–551. doi:10.1101/gr.3096505. ISSN 1088-9051. PMC 1074368 . PMID 15805494.
- ↑ Albà M, Tompa P, Veitia R (2007), Volff J (ed.), "Amino Acid Repeats and the Structure and Evolution of Proteins", Genome Dynamics, Basel: KARGER, 3: 119–130, doi:10.1159/000107607, ISBN 978-3-8055-8340-4, PMID 18753788 , retrieved 2020-11-03
- ↑ Marcotte EM, Pellegrini M, Yeates TO, Eisenberg D (1999-10-15). "A census of protein repeats". Journal of Molecular Biology. 293 (1): 151–160. doi:10.1006/jmbi.1999.3136. PMID 10512723.
- ↑ Marino SM, Gladyshev VN (2012-02-10). "Analysis and Functional Prediction of Reactive Cysteine Residues". Journal of Biological Chemistry. 287 (7): 4419–4425. doi: 10.1074/jbc.R111.275578 . ISSN 0021-9258. PMC 3281665 . PMID 22157013.
- ↑ Dorsman JC (2002-06-15). "Strong aggregation and increased toxicity of polyleucine over polyglutamine stretches in mammalian cells". Human Molecular Genetics. 11 (13): 1487–1496. doi: 10.1093/hmg/11.13.1487 . PMID 12045202.
- ↑ Oma Y, Kino Y, Sasagawa N, Ishiura S (2004-05-14). "Intracellular Localization of Homopolymeric Amino Acid-containing Proteins Expressed in Mammalian Cells". Journal of Biological Chemistry. 279 (20): 21217–21222. doi: 10.1074/jbc.M309887200 . ISSN 0021-9258. PMID 14993218. S2CID 23798438.
- ↑ Ellegren H (2004-06-01). "Microsatellites: simple sequences with complex evolution". Nature Reviews Genetics. 5 (6): 435–445. doi:10.1038/nrg1348. ISSN 1471-0056. PMID 15153996. S2CID 11975343.
- ↑ Verstrepen KJ, Jansen A, Lewitter F, Fink GR (2005-09-01). "Intragenic tandem repeats generate functional variability". Nature Genetics. 37 (9): 986–990. doi:10.1038/ng1618. ISSN 1061-4036. PMC 1462868 . PMID 16086015.
- ↑ Siwach P, Pophaly SD, Ganesh S (2006-07-01). "Genomic and Evolutionary Insights into Genes Encoding Proteins with Single Amino Acid Repeats". Molecular Biology and Evolution. 23 (7): 1357–1369. doi: 10.1093/molbev/msk022 . ISSN 1537-1719. PMID 16618963.
- ↑ Moxon R, Bayliss C, Hood D (2006-12-01). "Bacterial Contingency Loci: The Role of Simple Sequence DNA Repeats in Bacterial Adaptation". Annual Review of Genetics. 40 (1): 307–333. doi:10.1146/annurev.genet.40.110405.090442. ISSN 0066-4197. PMID 17094739.
- ↑ Toll-Riera M, Rado-Trilla N, Martys F, Alba MM (2012-03-01). "Role of Low-Complexity Sequences in the Formation of Novel Protein Coding Sequences". Molecular Biology and Evolution. 29 (3): 883–886. doi: 10.1093/molbev/msr263 . ISSN 0737-4038. PMID 22045997.
- ↑ Ohno S, Epplen JT (1983-06-01). "The primitive code and repeats of base oligomers as the primordial protein-encoding sequence". Proceedings of the National Academy of Sciences. 80 (11): 3391–3395. Bibcode:1983PNAS...80.3391O. doi: 10.1073/pnas.80.11.3391 . ISSN 0027-8424. PMC 394049 . PMID 6574491.
- 1 2 Trifonov EN (September 2009). "The origin of the genetic code and of the earliest oligopeptides". Research in Microbiology. 160 (7): 481–486. doi:10.1016/j.resmic.2009.05.004. PMID 19524038.
- ↑ Akashi H, Gojobori T (2002-03-19). "Metabolic efficiency and amino acid composition in the proteomes of Escherichia coli and Bacillus subtilis". Proceedings of the National Academy of Sciences. 99 (6): 3695–3700. Bibcode:2002PNAS...99.3695A. doi: 10.1073/pnas.062526999 . ISSN 0027-8424. PMC 122586 . PMID 11904428.
- ↑ Barton MD, Delneri D, Oliver SG, Rattray M, Bergman CM (2010-08-17). Bähler J (ed.). "Evolutionary Systems Biology of Amino Acid Biosynthetic Cost in Yeast". PLOS ONE. 5 (8): e11935. Bibcode:2010PLoSO...511935B. doi: 10.1371/journal.pone.0011935 . ISSN 1932-6203. PMC 2923148 . PMID 20808905.
- ↑ Radó-Trilla N, Albà M (2012). "Dissecting the role of low-complexity regions in the evolution of vertebrate proteins". BMC Evolutionary Biology. 12 (1): 155. Bibcode:2012BMCEE..12..155R. doi: 10.1186/1471-2148-12-155 . ISSN 1471-2148. PMC 3523016 . PMID 22920595.
- 1 2 Higgs PG, Pudritz RE (June 2009). "A Thermodynamic Basis for Prebiotic Amino Acid Synthesis and the Nature of the First Genetic Code". Astrobiology. 9 (5): 483–490. arXiv: 0904.0402 . Bibcode:2009AsBio...9..483H. doi:10.1089/ast.2008.0280. ISSN 1531-1074. PMID 19566427. S2CID 9039622.
- ↑ Trifonov E (2000-12-30). "Consensus temporal order of amino acids and evolution of the triplet code". Gene. 261 (1): 139–151. doi:10.1016/S0378-1119(00)00476-5. PMID 11164045.
- ↑ Trifonov EN (2004-08-01). "The Triplet Code From First Principles". Journal of Biomolecular Structure and Dynamics. 22 (1): 1–11. doi:10.1080/07391102.2004.10506975. ISSN 0739-1102. PMID 15214800. S2CID 28509952.
- ↑ Ferris JP, Hill AR, Liu R, Orgel LE (1996-05-02). "Synthesis of long prebiotic oligomers on mineral surfaces". Nature. 381 (6577): 59–61. Bibcode:1996Natur.381...59F. doi:10.1038/381059a0. hdl: 2060/19980119839 . ISSN 0028-0836. PMID 8609988. S2CID 4351826.
- ↑ Silva JM, Qi W, Pinho AJ, Pratas D (2022-12-28). "AlcoR: alignment-free simulation, mapping, and visualization of low-complexity regions in biological data". GigaScience. 12. doi:10.1093/gigascience/giad101. ISSN 2047-217X. PMC 10716826 . PMID 38091509.