Related Research Articles

Mass spectrometry (MS) is an analytical technique that is used to measure the mass-to-charge ratio of ions. The results are presented as a mass spectrum, a plot of intensity as a function of the mass-to-charge ratio. Mass spectrometry is used in many different fields and is applied to pure samples as well as complex mixtures.

Tandem mass spectrometry, also known as MS/MS or MS2, is a technique in instrumental analysis where two or more stages of analysis using one or more mass analyzer are performed with an additional reaction step in between these analyses to increase their abilities to analyse chemical samples. A common use of tandem MS is the analysis of biomolecules, such as proteins and peptides.

Gas chromatography–mass spectrometry (GC–MS) is an analytical method that combines the features of gas-chromatography and mass spectrometry to identify different substances within a test sample. Applications of GC–MS include drug detection, fire investigation, environmental analysis, explosives investigation, food and flavor analysis, and identification of unknown samples, including that of material samples obtained from planet Mars during probe missions as early as the 1970s. GC–MS can also be used in airport security to detect substances in luggage or on human beings. Additionally, it can identify trace elements in materials that were previously thought to have disintegrated beyond identification. Like liquid chromatography–mass spectrometry, it allows analysis and detection even of tiny amounts of a substance.
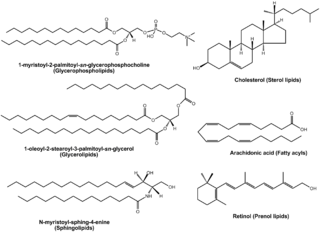
Lipidomics is the large-scale study of pathways and networks of cellular lipids in biological systems The word "lipidome" is used to describe the complete lipid profile within a cell, tissue, organism, or ecosystem and is a subset of the "metabolome" which also includes other major classes of biological molecules. Lipidomics is a relatively recent research field that has been driven by rapid advances in technologies such as mass spectrometry (MS), nuclear magnetic resonance (NMR) spectroscopy, fluorescence spectroscopy, dual polarisation interferometry and computational methods, coupled with the recognition of the role of lipids in many metabolic diseases such as obesity, atherosclerosis, stroke, hypertension and diabetes. This rapidly expanding field complements the huge progress made in genomics and proteomics, all of which constitute the family of systems biology.

Metabolomics is the scientific study of chemical processes involving metabolites, the small molecule substrates, intermediates, and products of cell metabolism. Specifically, metabolomics is the "systematic study of the unique chemical fingerprints that specific cellular processes leave behind", the study of their small-molecule metabolite profiles. The metabolome represents the complete set of metabolites in a biological cell, tissue, organ, or organism, which are the end products of cellular processes. Messenger RNA (mRNA), gene expression data, and proteomic analyses reveal the set of gene products being produced in the cell, data that represents one aspect of cellular function. Conversely, metabolic profiling can give an instantaneous snapshot of the physiology of that cell, and thus, metabolomics provides a direct "functional readout of the physiological state" of an organism. There are indeed quantifiable correlations between the metabolome and the other cellular ensembles, which can be used to predict metabolite abundances in biological samples from, for example mRNA abundances. One of the ultimate challenges of systems biology is to integrate metabolomics with all other -omics information to provide a better understanding of cellular biology.

The metabolome refers to the complete set of small-molecule chemicals found within a biological sample. The biological sample can be a cell, a cellular organelle, an organ, a tissue, a tissue extract, a biofluid or an entire organism. The small molecule chemicals found in a given metabolome may include both endogenous metabolites that are naturally produced by an organism as well as exogenous chemicals that are not naturally produced by an organism.
Infrared multiple photon dissociation (IRMPD) is a technique used in mass spectrometry to fragment molecules in the gas phase usually for structural analysis of the original (parent) molecule.

Liquid chromatography–mass spectrometry (LC–MS) is an analytical chemistry technique that combines the physical separation capabilities of liquid chromatography with the mass analysis capabilities of mass spectrometry (MS). Coupled chromatography – MS systems are popular in chemical analysis because the individual capabilities of each technique are enhanced synergistically. While liquid chromatography separates mixtures with multiple components, mass spectrometry provides spectral information that may help to identify each separated component. MS is not only sensitive, but provides selective detection, relieving the need for complete chromatographic separation. LC–MS is also appropriate for metabolomics because of its good coverage of a wide range of chemicals. This tandem technique can be used to analyze biochemical, organic, and inorganic compounds commonly found in complex samples of environmental and biological origin. Therefore, LC–MS may be applied in a wide range of sectors including biotechnology, environment monitoring, food processing, and pharmaceutical, agrochemical, and cosmetic industries. Since the early 2000s, LC–MS has also begun to be used in clinical applications.

Protein mass spectrometry refers to the application of mass spectrometry to the study of proteins. Mass spectrometry is an important method for the accurate mass determination and characterization of proteins, and a variety of methods and instrumentations have been developed for its many uses. Its applications include the identification of proteins and their post-translational modifications, the elucidation of protein complexes, their subunits and functional interactions, as well as the global measurement of proteins in proteomics. It can also be used to localize proteins to the various organelles, and determine the interactions between different proteins as well as with membrane lipids.

In mass spectrometry, fragmentation is the dissociation of energetically unstable molecular ions formed from passing the molecules mass spectrum. These reactions are well documented over the decades and fragmentation patterns are useful to determine the molar weight and structural information of unknown molecules. Fragmentation that occurs in tandem mass spectrometry experiments has been a recent focus of research, because this data helps facilitate the identification of molecules.
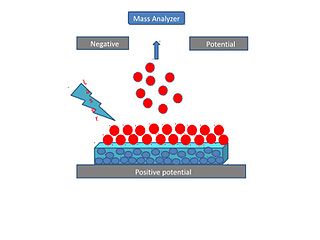
Surface-assisted laser desorption/ionization (SALDI) is a soft laser desorption technique used for mass spectrometry analysis of biomolecules, polymers, and small organic molecules. In its first embodiment Koichi Tanaka used a cobalt/glycerol liquid matrix and subsequent applications included a graphite/glycerol liquid matrix as well as a solid surface of porous silicon. The porous silicon represents the first matrix-free SALDI surface analysis allowing for facile detection of intact molecular ions, these porous silicon surfaces also facilitated the analysis of small molecules at the yoctomole level. At present laser desorption/ionization methods using other inorganic matrices such as nanomaterials are often regarded as SALDI variants. As an example, silicon nanowires as well as Titania nanotube arrays (NTA) have been used as substrates to detect small molecules. SALDI is used to detect proteins and protein-protein complexes. A related method named "ambient SALDI" - which is a combination of conventional SALDI with ambient mass spectrometry incorporating the direct analysis real time (DART) ion source has also been demonstrated. SALDI is considered one of the most important techniques in MS and has many applications.
The Human Metabolome Database (HMDB) is a comprehensive, high-quality, freely accessible, online database of small molecule metabolites found in the human body. It has been created by the Human Metabolome Project funded by Genome Canada and is one of the first dedicated metabolomics databases. The HMDB facilitates human metabolomics research, including the identification and characterization of human metabolites using NMR spectroscopy, GC-MS spectrometry and LC/MS spectrometry. To aid in this discovery process, the HMDB contains three kinds of data: 1) chemical data, 2) clinical data, and 3) molecular biology/biochemistry data (Fig. 1–3). The chemical data includes 41,514 metabolite structures with detailed descriptions along with nearly 10,000 NMR, GC-MS and LC/MS spectra.

In the field of cellular biology, single-cell analysis and subcellular analysis is the study of genomics, transcriptomics, proteomics, metabolomics and cell–cell interactions at the single cell level. The concept of single-cell analysis originated in the 1970s. Before the discovery of heterogeneity, single-cell analysis mainly referred to the analysis or manipulation of an individual cell in a bulk population of cells at a particular condition using optical or electronic microscope. To date, due to the heterogeneity seen in both eukaryotic and prokaryotic cell populations, analyzing a single cell makes it possible to discover mechanisms not seen when studying a bulk population of cells. Technologies such as fluorescence-activated cell sorting (FACS) allow the precise isolation of selected single cells from complex samples, while high throughput single cell partitioning technologies, enable the simultaneous molecular analysis of hundreds or thousands of single unsorted cells; this is particularly useful for the analysis of transcriptome variation in genotypically identical cells, allowing the definition of otherwise undetectable cell subtypes. The development of new technologies is increasing our ability to analyze the genome and transcriptome of single cells, as well as to quantify their proteome and metabolome. Mass spectrometry techniques have become important analytical tools for proteomic and metabolomic analysis of single cells. Recent advances have enabled quantifying thousands of protein across hundreds of single cells, and thus make possible new types of analysis. In situ sequencing and fluorescence in situ hybridization (FISH) do not require that cells be isolated and are increasingly being used for analysis of tissues.
The Yeast Metabolome Database (YMDB) is a comprehensive, high-quality, freely accessible, online database of small molecule metabolites found in or produced by Saccharomyces cerevisiae. The YMDB was designed to facilitate yeast metabolomics research, specifically in the areas of general fermentation as well as wine, beer and fermented food analysis. YMDB supports the identification and characterization of yeast metabolites using NMR spectroscopy, GC-MS spectrometry and Liquid chromatography–mass spectrometry. The YMDB contains two kinds of data: 1) chemical data and 2) molecular biology/biochemistry data. The chemical data includes 2027 metabolite structures with detailed metabolite descriptions along with nearly 4000 NMR, GC-MS and LC/MS spectra.

Desorption/ionization on silicon (DIOS) is a soft laser desorption method used to generate gas-phase ions for mass spectrometry analysis. DIOS is considered the first surface-based surface-assisted laser desorption/ionization (SALDI-MS) approach. Prior approaches were accomplished using nanoparticles in a matrix of glycerol, while DIOS is a matrix-free technique in which a sample is deposited on a nanostructured surface and the sample desorbed directly from the nanostructured surface through the adsorption of laser light energy. DIOS has been used to analyze organic molecules, metabolites, biomolecules and peptides, and, ultimately, to image tissues and cells.

Gary Siuzdak is an American chemist best known for his work in the field of metabolomics, activity metabolomics, and mass spectrometry. His lab discovered indole-3-propionic acid as a gut bacteria derived metabolite in 2009. He is currently the Professor and Director of The Center for Metabolomics and Mass Spectrometry at Scripps Research in La Jolla, California. Siuzdak has also made contributions to virus analysis, viral structural dynamics, as well as developing mass spectrometry imaging technology using nanostructured surfaces. The Siuzdak lab is also responsible for creating the research tools eXtensible Computational Mass Spectrometry (XCMS), METLIN, METLIN Neutral Loss and Q-MRM. As of January 2021, the XCMS/METLIN platform has over 50,000 registered users.

XCMS Online is a cloud version of the original eXtensible Computational Mass Spectrometry (XCMS) technology, created by the Siuzdak Lab at Scripps Research. XCMS introduced the concept of nonlinear retention time alignment that allowed for the statistical assessment of the detected peaks across LCMS and GCMS datasets. XCMS Online was designed to facilitate XCMS analyses through a cloud portal and as a more straightforward way to analyze, visualize and share untargeted metabolomic data. Further to this, the combination of XCMS and METLIN allows for the identification of known molecules using METLIN's tandem mass spectrometry data, and enables the identification of unknown via similarity searching of tandem mass spectrometry data. XCMS Online has also become a systems biology tool for integrating different omic data sets. As of January 2021, the XCMSOnline - METLIN platform has over 44,000 registered users. XCMS - METLIN was recognized in 2023 as the year's top analytical innovation.

Emma Schymanski is chemist known for her work identifying unknown organic compounds, particularly pollutants, and is an advocate for open science.
SIRIUS is a Java-based open-source software for the identification of small molecules from fragmentation mass spectrometry data without the use of spectral libraries. It combines the analysis of isotope patterns in MS1 spectra with the analysis of fragmentation patterns in MS2 spectra. SIRIUS is the umbrella application comprising CSI:FingerID, CANOPUS, COSMIC and ZODIAC.
References
- ↑ Xue, Jingchuan; Guijas, Carlos; Benton, H. Paul; Warth, Benedikt; Siuzdak, Gary (October 2020). "METLIN MS 2 molecular standards database: a broad chemical and biological resource". Nature Methods. 17 (10): 953–954. doi:10.1038/s41592-020-0942-5. ISSN 1548-7105. PMC 8802982 . PMID 32839599.
- 1 2 Smith CA, O'Maille G, Want EJ, Qin C, Trauger SA, Brandon TR, et al. (December 2005). "METLIN: a metabolite mass spectral database". Therapeutic Drug Monitoring. 27 (6): 747–51. doi:10.1097/01.ftd.0000179845.53213.39. PMID 16404815. S2CID 14774455.
- 1 2 3 Guijas C, Montenegro-Burke JR, Domingo-Almenara X, Palermo A, Warth B, Hermann G, et al. (March 2018). "METLIN: A Technology Platform for Identifying Knowns and Unknowns". Analytical Chemistry. 90 (5): 3156–3164. doi:10.1021/acs.analchem.7b04424. PMC 5933435 . PMID 29381867.
- ↑ Hoang, Corey; Uritboonthai, Winnie; Hoang, Linh; Billings, Elizabeth M.; Aisporna, Aries; Nia, Farshad A.; Derks, Rico J. E.; Williamson, James R.; Giera, Martin; Siuzdak, Gary (2024-03-26). "Tandem Mass Spectrometry across Platforms". Analytical Chemistry. doi:10.1021/acs.analchem.3c05576. ISSN 0003-2700.
- 1 2 Aisporna, Aries; Benton, H. Paul; Chen, Andy; Derks, Rico J. E.; Galano, Jean Marie; Giera, Martin; Siuzdak, Gary (2022-03-02). "Neutral Loss Mass Spectral Data Enhances Molecular Similarity Analysis in METLIN". Journal of the American Society for Mass Spectrometry. 33 (3): 530–534. doi:10.1021/jasms.1c00343. ISSN 1044-0305. PMC 10131246 . PMID 35174708. S2CID 246902725.
- ↑ Giera, Martin; Yanes, Oscar; Siuzdak, Gary (2022-01-04). "Metabolite discovery: Biochemistry's scientific driver". Cell Metabolism. 34 (1): 21–34. doi: 10.1016/j.cmet.2021.11.005 . hdl: 1887/3250578 . ISSN 1550-4131. PMID 34986335. S2CID 245729571.
- 1 2 3 4 5 Xue J, Guijas C, Benton HP, Warth B, Siuzdak G (August 2020). "2 molecular standards database: a broad chemical and biological resource". Nature Methods. 17 (10): 953–954. doi:10.1038/s41592-020-0942-5. PMC 8802982 . PMID 32839599.
- ↑ Guijas, Carlos; To, Andrew; Montenegro-Burke, J. Rafael; Domingo-Almenara, Xavier; Alipio-Gloria, Zaida; Kok, Bernard P.; Saez, Enrique; Alvarez, Nicole H.; Johnson, Kristen A.; Siuzdak, Gary (August 2022). "Drug-Initiated Activity Metabolomics Identifies Myristoylglycine as a Potent Endogenous Metabolite for Human Brown Fat Differentiation". Metabolites. 12 (8): 749. doi: 10.3390/metabo12080749 . ISSN 2218-1989. PMC 9415469 . PMID 36005620.
- 1 2 "The Analytical Scientist Innovation Awards 2023". The Analytical Scientist. 2023-12-12. Retrieved 2023-12-14.
- ↑ Heim, Wilasinee; Aisporna, Aries; Hoang, Linh; Benton, H. Paul; Siuzdak, Gary (2023), Ivanisevic, Julijana; Giera, Martin (eds.), "METLIN Tandem Mass Spectrometry and Neutral Loss Databases for the Identification of Microbial Natural Products and Other Chemical Entities", A Practical Guide to Metabolomics Applications in Health and Disease: From Samples to Insights into Metabolism, Cham: Springer International Publishing, pp. 105–124, doi:10.1007/978-3-031-44256-8_5, ISBN 978-3-031-44256-8 , retrieved 2024-03-27
- 1 2 Benton HP, Wong DM, Trauger SA, Siuzdak G (August 2008). "XCMS2: processing tandem mass spectrometry data for metabolite identification and structural characterization". Analytical Chemistry. 80 (16): 6382–9. doi:10.1021/ac800795f. PMC 2728033 . PMID 18627180.
- 1 2 Tautenhahn R, Cho K, Uritboonthai W, Zhu Z, Patti GJ, Siuzdak G (September 2012). "An accelerated workflow for untargeted metabolomics using the METLIN database". Nature Biotechnology. 30 (9): 826–8. doi:10.1038/nbt.2348. PMC 3666346 . PMID 22965049.
- 1 2 Domingo-Almenara, Xavier; Montenegro-Burke, J. Rafael; Ivanisevic, Julijana; Thomas, Aurelien; Sidibé, Jonathan; Teav, Tony; Guijas, Carlos; Aisporna, Aries E.; Rinehart, Duane; Hoang, Linh; Nordström, Anders (September 2018). "XCMS-MRM and METLIN-MRM: a cloud library and public resource for targeted analysis of small molecules". Nature Methods. 15 (9): 681–684. doi:10.1038/s41592-018-0110-3. ISSN 1548-7105. PMC 6629029 . PMID 30150755.
- ↑ Majumder, Erica L.-W.; Billings, Elizabeth M.; Benton, H. Paul; Martin, Richard L.; Palermo, Amelia; Guijas, Carlos; Rinschen, Markus M.; Domingo-Almenara, Xavier; Montenegro-Burke, J. Rafael; Tagtow, Bradley A.; Plumb, Robert S. (2021-01-22). "Cognitive analysis of metabolomics data for systems biology". Nature Protocols. 16 (3): 1376–1418. doi:10.1038/s41596-020-00455-4. ISSN 1754-2189. OSTI 1774918. PMC 10357461 . PMID 33483720. S2CID 231687415.
- ↑ Heim, Wilasinee; Aisporna, Aries; Hoang, Linh; Benton, H. Paul; Siuzdak, Gary (2023), Ivanisevic, Julijana; Giera, Martin (eds.), "METLIN Tandem Mass Spectrometry and Neutral Loss Databases for the Identification of Microbial Natural Products and Other Chemical Entities", A Practical Guide to Metabolomics Applications in Health and Disease: From Samples to Insights into Metabolism, Learning Materials in Biosciences, Cham: Springer International Publishing, pp. 105–124, doi:10.1007/978-3-031-44256-8_5, ISBN 978-3-031-44256-8 , retrieved 2024-03-01