Phenylketonuria (PKU) is an inborn error of metabolism that results in decreased metabolism of the amino acid phenylalanine. Untreated PKU can lead to intellectual disability, seizures, behavioral problems, and mental disorders. It may also result in a musty smell and lighter skin. A baby born to a mother who has poorly treated PKU may have heart problems, a small head, and low birth weight.

Congenital adrenal hyperplasia (CAH) is a group of autosomal recessive disorders characterized by impaired cortisol synthesis. It results from the deficiency of one of the five enzymes required for the synthesis of cortisol in the adrenal cortex. Most of these disorders involve excessive or deficient production of hormones such as glucocorticoids, mineralocorticoids, or sex steroids, and can alter development of primary or secondary sex characteristics in some affected infants, children, or adults. It is one of the most common autosomal recessive disorders in humans.

Galactosemia is a rare genetic metabolic disorder that affects an individual's ability to metabolize the sugar galactose properly. Galactosemia follows an autosomal recessive mode of inheritance that confers a deficiency in an enzyme responsible for adequate galactose degradation.

Newborn screening (NBS) is a public health program of screening in infants shortly after birth for conditions that are treatable, but not clinically evident in the newborn period. The goal is to identify infants at risk for these conditions early enough to confirm the diagnosis and provide intervention that will alter the clinical course of the disease and prevent or ameliorate the clinical manifestations. NBS started with the discovery that the amino acid disorder phenylketonuria (PKU) could be treated by dietary adjustment, and that early intervention was required for the best outcome. Infants with PKU appear normal at birth, but are unable to metabolize the essential amino acid phenylalanine, resulting in irreversible intellectual disability. In the 1960s, Robert Guthrie developed a simple method using a bacterial inhibition assay that could detect high levels of phenylalanine in blood shortly after a baby was born. Guthrie also pioneered the collection of blood on filter paper which could be easily transported, recognizing the need for a simple system if the screening was going to be done on a large scale. Newborn screening around the world is still done using similar filter paper. NBS was first introduced as a public health program in the United States in the early 1960s, and has expanded to countries around the world.
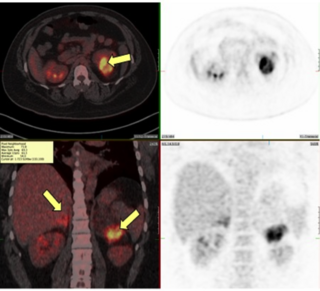
Congenital adrenal hyperplasia due to 21-hydroxylase deficiency (CAH) is a genetic disorder characterized by impaired production of cortisol in the adrenal glands.
Inborn errors of metabolism form a large class of genetic diseases involving congenital disorders of enzyme activities. The majority are due to defects of single genes that code for enzymes that facilitate conversion of various substances (substrates) into others (products). In most of the disorders, problems arise due to accumulation of substances which are toxic or interfere with normal function, or due to the effects of reduced ability to synthesize essential compounds. Inborn errors of metabolism are often referred to as congenital metabolic diseases or inherited metabolic disorders. Another term used to describe these disorders is "enzymopathies". This term was created following the study of biodynamic enzymology, a science based on the study of the enzymes and their products. Finally, inborn errors of metabolism were studied for the first time by British physician Archibald Garrod (1857–1936), in 1908. He is known for work that prefigured the "one gene-one enzyme" hypothesis, based on his studies on the nature and inheritance of alkaptonuria. His seminal text, Inborn Errors of Metabolism, was published in 1923.
Ivar Asbjørn Følling was a Norwegian physician and biochemist. He first described the disease commonly known as Følling's disease or phenylketonuria (PKU).

Group B streptococcal infection, also known as Group B streptococcal disease or just Group B strepinfection,

The following outline is provided as an overview of and topical guide to biochemistry:

Tetrahydrobiopterin deficiency (THBD, BH4D) is a rare metabolic disorder that increases the blood levels of phenylalanine. Phenylalanine is an amino acid obtained normally through the diet, but can be harmful if excess levels build up, causing intellectual disability and other serious health problems. In healthy individuals, it is metabolised (hydroxylated) into tyrosine, another amino acid, by phenylalanine hydroxylase. However, this enzyme requires tetrahydrobiopterin as a cofactor and thus its deficiency slows phenylalanine metabolism.

6-Pyruvoyltetrahydropterin synthase deficiency is an autosomal recessive disorder that causes malignant hyperphenylalaninemia due to tetrahydrobiopterin deficiency. It is a recessive disorder that is accompanied by hyperphenylalaninemia. Commonly reported symptoms are initial truncal hypotonia, subsequent appendicular hypertonia, bradykinesia, cogwheel rigidity, generalized dystonia, and marked diurnal fluctuation. Other reported clinical features include difficulty in swallowing, oculogyric crises, somnolence, irritability, hyperthermia, and seizures. Chorea, athetosis, hypersalivation, rash with eczema, and sudden death have also been reported. Patients with mild phenotypes may deteriorate if given folate antagonists such as methotrexate, which can interfere with a salvage pathway through which dihydrobiopterin is converted into tetrahydrobiopterin via dihydrofolate reductase. Treatment options include substitution with neurotransmitter precursors, monoamine oxidase inhibitors, and tetrahydrobiopterin. Response to treatment is variable and the long-term and functional outcome is unknown. To provide a basis for improving the understanding of the epidemiology, genotype–phenotype correlation and outcome of these diseases, their impact on the quality of life of patients, and for evaluating diagnostic and therapeutic strategies a patient registry was established by the noncommercial International Working Group on Neurotransmitter Related Disorders (iNTD).
Robert Guthrie, MD, Ph.D. was an American microbiologist, best known for developing the bacterial inhibition assay used to screen infants for phenylketonuria at birth, before the development of irreversible neurological damage. Guthrie also pioneered the collection of whole blood on specially designed filter paper, commonly known as "Guthrie cards" as a sample medium that could be easily collected, transported and tested. Although Guthrie is best known for developing the test for phenylketonuria, he worked tirelessly to raise awareness of the need to screen for treatable conditions and adapted his method to early screening tests for galactosemia and maple syrup urine disease.
Measurement of immunoreactive trypsinogen (IRT) in blood of newborn babies is an assay in rapidly increasing use as a screening test for cystic fibrosis (CF).
Galactose-1-phosphate uridylyltransferase deficiency(classic galactosemia) is the most common type of galactosemia, an inborn error of galactose metabolism, caused by a deficiency of the enzyme galactose-1-phosphate uridylyltransferase. It is an autosomal recessive metabolic disorder that can cause liver disease and death if untreated. Treatment of galactosemia is most successful if initiated early and includes dietary restriction of lactose intake. Because early intervention is key, galactosemia is included in newborn screening programs in many areas. On initial screening, which often involves measuring the concentration of galactose in blood, classic galactosemia may be indistinguishable from other inborn errors of galactose metabolism, including galactokinase deficiency and galactose epimerase deficiency. Further analysis of metabolites and enzyme activities are needed to identify the specific metabolic error.

A urine test strip or dipstick is a basic diagnostic tool used to determine pathological changes in a patient's urine in standard urinalysis.
Hyperphenylalaninemia is a medical condition characterized by mildly or strongly elevated concentrations of the amino acid phenylalanine in the blood. Phenylketonuria (PKU) can result in severe hyperphenylalaninemia. Phenylalanine concentrations are routinely screened in newborns by the neonatal heel prick, which takes a few drops of blood from the heel of the infant. Standard phenylalanine concentrations in unaffected persons are about 2-6mg/dl phenylalanine concentrations in those with untreated hyperphenylalaninemia can be up to 20 mg/dL. Measurable IQ deficits are often detected as phenylalanine levels approach 10 mg/dL. Phenylketonuria (PKU)-like symptoms, including more pronounced developmental defects, skin irritation, and vomiting, may appear when phenylalanine levels are near 20 mg/dL .Hyperphenylalaninemia is a recessive hereditary metabolic disorder that is caused by the body's failure to convert phenylalanine to tyrosine as a result of the entire or partial absence of the enzyme phenylalanine hydroxylase.

The Newborn Screening Saves Lives Reauthorization Act of 2014 is a bill that would amend the Public Health Service Act to reauthorize grant programs and other initiatives to promote expanded screening of newborns and children for heritable disorders.

Louis Isaac Woolf was a British biochemist who played a crucial role in the early detection and the treatment of phenylketonuria (PKU).