
Huntington's disease (HD), also known as Huntington's chorea, is an incurable neurodegenerative disease that is mostly inherited. The earliest symptoms are often subtle problems with mood or mental/psychiatric abilities. A general lack of coordination and an unsteady gait often follow. It is also a basal ganglia disease causing a hyperkinetic movement disorder known as chorea. As the disease advances, uncoordinated, involuntary body movements of chorea become more apparent. Physical abilities gradually worsen until coordinated movement becomes difficult and the person is unable to talk. Mental abilities generally decline into dementia, depression, apathy, and impulsivity at times. The specific symptoms vary somewhat between people. Symptoms usually begin between 30 and 50 years of age, and can start at any age but are usually seen around the age of 40. The disease may develop earlier in each successive generation. About eight percent of cases start before the age of 20 years, and are known as juvenile HD, which typically present with the slow movement symptoms of Parkinson's disease rather than those of chorea.

Wilson's disease is a genetic disorder in which excess copper builds up in the body. Symptoms are typically related to the brain and liver. Liver-related symptoms include vomiting, weakness, fluid build-up in the abdomen, swelling of the legs, yellowish skin, and itchiness. Brain-related symptoms include tremors, muscle stiffness, trouble in speaking, personality changes, anxiety, and psychosis.

Leigh syndrome is an inherited neurometabolic disorder that affects the central nervous system. It is named after Archibald Denis Leigh, a British neuropsychiatrist who first described the condition in 1951. Normal levels of thiamine, thiamine monophosphate, and thiamine diphosphate are commonly found, but there is a reduced or absent level of thiamine triphosphate. This is thought to be caused by a blockage in the enzyme thiamine-diphosphate kinase, and therefore treatment in some patients would be to take thiamine triphosphate daily. While the majority of patients typically exhibit symptoms between the ages of 3 and 12 months, instances of adult onset have also been documented.

Chorea-acanthocytosis is a rare hereditary disease caused by a mutation in a gene that directs structural proteins in red blood cells. It belongs to a group of four diseases characterized under the name neuroacanthocytosis. When a patient's blood is viewed under a microscope, some of the red blood cells appear thorny. These thorny cells are called acanthocytes.
Pantothenate kinase-associated neurodegeneration (PKAN), formerly called Hallervorden–Spatz syndrome, is a genetic degenerative disease of the brain that can lead to parkinsonism, dystonia, dementia, and ultimately death. Neurodegeneration in PKAN is accompanied by an excess of iron that progressively builds up in the brain.

Aceruloplasminemia is a rare autosomal recessive disorder in which the liver can not synthesize the protein ceruloplasmin properly, which is needed to transport copper around the blood. Copper deficiency in the brain results in neurological problems that generally appear in adulthood and worsen over time. .
Neuroacanthocytosis is a label applied to several genetic neurological conditions in which the blood contains misshapen, spiculated red blood cells called acanthocytes.
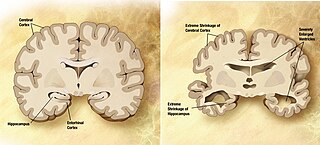
A neurodegenerative disease is caused by the progressive loss of structure or function of neurons, in the process known as neurodegeneration. Such neuronal damage may ultimately involve cell death. Neurodegenerative diseases include amyotrophic lateral sclerosis, multiple sclerosis, Parkinson's disease, Alzheimer's disease, Huntington's disease, multiple system atrophy, tauopathies, and prion diseases. Neurodegeneration can be found in the brain at many different levels of neuronal circuitry, ranging from molecular to systemic. Because there is no known way to reverse the progressive degeneration of neurons, these diseases are considered to be incurable; however research has shown that the two major contributing factors to neurodegeneration are oxidative stress and inflammation. Biomedical research has revealed many similarities between these diseases at the subcellular level, including atypical protein assemblies and induced cell death. These similarities suggest that therapeutic advances against one neurodegenerative disease might ameliorate other diseases as well.

Infantile neuroaxonal dystrophy (INAD) is a rare pervasive developmental disorder that primarily affects the nervous system. Individuals with infantile neuroaxonal dystrophy typically do not have any symptoms at birth, but between the ages of about 6 and 18 months they begin to experience delays in acquiring new motor and intellectual skills, such as crawling or beginning to speak. Eventually they lose previously acquired skills.

85 kDa calcium-independent phospholipase A2, also known as 85/88 kDa calcium-independent phospholipase A2, Group VI phospholipase A2, Intracellular membrane-associated calcium-independent phospholipase A2 beta, or Patatin-like phospholipase domain-containing protein 9 is an enzyme that in humans is encoded by the PLA2G6 gene.
Leukoencephalopathy with neuroaxonal spheroids (LENAS) is an extremely rare kind of leukoencephalopathy and is classified as a neurodegenerative disease. LENAS is a cause of severe and subacute dementia that results from damage to certain areas of the brain. This damage is to a type of brain tissue called white matter and axon damage due to swellings which are termed spheroids.
Nervous system diseases, also known as nervous system or neurological disorders, refers to a small class of medical conditions affecting the nervous system. This category encompasses over 600 different conditions, including genetic disorders, infections, cancer, seizure disorders, conditions with a cardiovascular origin, congenital and developmental disorders, and degenerative disorders.
Myoclonic dystonia or Myoclonus dystonia syndrome is a rare movement disorder that induces spontaneous muscle contraction causing abnormal posture. The prevalence of myoclonus dystonia has not been reported, however, this disorder falls under the umbrella of movement disorders which affect thousands worldwide. Myoclonus dystonia results from mutations in the SGCE gene coding for an integral membrane protein found in both neurons and muscle fibers. Those suffering from this disease exhibit symptoms of rapid, jerky movements of the upper limbs (myoclonus), as well as distortion of the body's orientation due to simultaneous activation of agonist and antagonist muscles (dystonia).

Basal ganglia disease is a group of physical problems that occur when the group of nuclei in the brain known as the basal ganglia fail to properly suppress unwanted movements or to properly prime upper motor neuron circuits to initiate motor function. Research indicates that increased output of the basal ganglia inhibits thalamocortical projection neurons. Proper activation or deactivation of these neurons is an integral component for proper movement. If something causes too much basal ganglia output, then the ventral anterior (VA) and ventral lateral (VL) thalamocortical projection neurons become too inhibited, and one cannot initiate voluntary movement. These disorders are known as hypokinetic disorders. However, a disorder leading to abnormally low output of the basal ganglia leads to reduced inhibition, and thus excitation, of the thalamocortical projection neurons which synapse onto the cortex. This situation leads to an inability to suppress unwanted movements. These disorders are known as hyperkinetic disorders.
Kufor–Rakeb syndrome (KRS) is an autosomal recessive disorder of juvenile onset also known as Parkinson disease-9 (PARK9). It is named after Kufr Rakeb in Irbid, Jordan. Kufor–Rakeb syndrome was first identified in this region in Jordan with a Jordanian couple's 5 children who had rigidity, mask-like face, and bradykinesia. The disease was first described in 1994 by Najim Al-Din et al. The OMIM number is 606693.
Neurodegeneration with brain iron accumulation is a heterogenous group of inherited neurodegenerative diseases, still under research, in which iron accumulates in the basal ganglia, either resulting in progressive dystonia, parkinsonism, spasticity, optic atrophy, retinal degeneration, neuropsychiatric, or diverse neurologic abnormalities. Some of the NBIA disorders have also been associated with several genes in synapse and lipid metabolism related pathways. NBIA is not one disease but an entire group of disorders, characterized by an accumulation of brain iron, sometimes in the presence of axonal spheroids in the central nervous system.
Benign hereditary chorea (BHC), also known as benign familial chorea, is a rare autosomal dominant neurogenetic syndrome. It typically presents itself in childhood with isolated chorea, with average to below average intelligence. Unlike other neurogenetic causes of chorea such as Huntington's disease, BHC is not progressive, and not associated with cognitive decline or psychiatric problems in the vast majority of cases.

The HFE H63D is a single-nucleotide polymorphism in the HFE gene, which results in the substitution of a histidine for an aspartic acid at amino acid position 63 of the HFE protein (p.His63Asp). HFE participates in the regulation of iron absorption.
Mitochondrial membrane protein-associated neurodegeneration (MPAN) is a genetic neurodegenerative disease that causes dystonia, parkinsonism, and iron accumulation in the brain. It is caused by mutations to the gene C19orf12, which has unknown function. This was originally discovered as an autosomal recessive disorder, caused by individuals having two mutations to the gene C19orf12, but autosomal dominant disease caused by a single mutation in the same gene has also been rarely described. Due to the common features of neurodegeneration, brain iron accumulation, and movement disorder it is classified as a neurodegeneration with brain iron accumulation (NBIA) disorder and another name for the condition is neurodegeneration with brain iron accumulation 4 (NBIA4).
Biotin-thiamine-responsive basal ganglia disease (BTBGD) is a rare disease that affects the nervous system, particularly the basal ganglia in the brain. It is a treatable neurometabolic disorder with autosomal recessive inheritance. First described in 1998 and then genetically distinguished in 2005, the disease is characterized by progressive brain damage that, if left untreated, can lead to coma and/or death. Commonly observed in individuals with BTBGD is recurring subacute encephalopathy along with confusion, seizures, and disordered movement (hypokinesia).


