
Elias James Corey is an American organic chemist. In 1990, he won the Nobel Prize in Chemistry "for his development of the theory and methodology of organic synthesis", specifically retrosynthetic analysis. Regarded by many as one of the greatest living chemists, he has developed numerous synthetic reagents, methodologies and total syntheses and has advanced the science of organic synthesis considerably.

The aldol reaction is a means of forming carbon–carbon bonds in organic chemistry. Discovered independently by the Russian chemist Alexander Borodin in 1869 and by the French chemist Charles-Adolphe Wurtz in 1872, the reaction combines two carbonyl compounds to form a new β-hydroxy carbonyl compound. These products are known as aldols, from the aldehyde + alcohol, a structural motif seen in many of the products. Aldol structural units are found in many important molecules, whether naturally occurring or synthetic. For example, the aldol reaction has been used in the large-scale production of the commodity chemical pentaerythritol and the synthesis of the heart disease drug Lipitor.

Organolithium reagents are organometallic compounds that contain carbon–lithium bonds. These reagents are important in organic synthesis, and are frequently used to transfer the organic group or the lithium atom to the substrates in synthetic steps, through nucleophilic addition or simple deprotonation. Organolithium reagents are used in industry as an initiator for anionic polymerization, which leads to the production of various elastomers. They have also been applied in asymmetric synthesis in the pharmaceutical industry. Due to the large difference in electronegativity between the carbon atom and the lithium atom, the C−Li bond is highly ionic. Owing to the polar nature of the C−Li bond, organolithium reagents are good nucleophiles and strong bases. For laboratory organic synthesis, many organolithium reagents are commercially available in solution form. These reagents are highly reactive, and are sometimes pyrophoric.
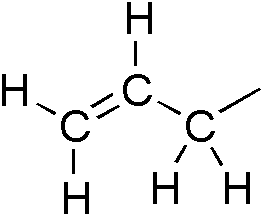
An allyl group is a substituent with the structural formula H2C=CH−CH2R, where R is the rest of the molecule. It consists of a methylene bridge (−CH2−) attached to a vinyl group (−CH=CH2). The name is derived from the Latin word for garlic, Allium sativum. In 1844, Theodor Wertheim isolated an allyl derivative from garlic oil and named it "Schwefelallyl". The term allyl applies to many compounds related to H2C=CH−CH2, some of which are of practical or of everyday importance, for example, allyl chloride. Allylation is any chemical reaction that adds an allyl group to a substrate.

The ene reaction is a chemical reaction between an alkene with an allylic hydrogen and a compound containing a multiple bond, in order to form a new σ-bond with migration of the ene double bond and 1,5 hydrogen shift. The product is a substituted alkene with the double bond shifted to the allylic position.

A chiral auxiliary is a stereogenic group or unit that is temporarily incorporated into an organic compound in order to control the stereochemical outcome of the synthesis. The chirality present in the auxiliary can bias the stereoselectivity of one or more subsequent reactions. The auxiliary can then be typically recovered for future use.

The Petasis reaction is the multi-component reaction of an amine, a carbonyl, and a vinyl- or aryl-boronic acid to form substituted amines.

In stereochemistry, asymmetric induction describes the preferential formation in a chemical reaction of one enantiomer or diastereoisomer over the other as a result of the influence of a chiral feature present in the substrate, reagent, catalyst or environment. Asymmetric induction is a key element in asymmetric synthesis.

The Stork enamine alkylation involves the addition of an enamine to a Michael acceptor or another electrophilic alkylation reagent to give an alkylated iminium product, which is hydrolyzed by dilute aqueous acid to give the alkylated ketone or aldehyde. Since enamines are generally produced from ketones or aldehydes, this overall process constitutes a selective monoalkylation of a ketone or aldehyde, a process that may be difficult to achieve directly.

Organoindium chemistry is the chemistry of compounds containing In-C bonds. The main application of organoindium chemistry is in the preparation of semiconducting components for microelectronic applications. The area is also of some interest in organic synthesis. Most organoindium compounds feature the In(III) oxidation state, akin to its lighter congeners Ga(III) and B(III).
The α-ketol rearrangement is the acid-, base-, or heat-induced 1,2-migration of an alkyl or aryl group in an α-hydroxy ketone or aldehyde to give an isomeric product.
The Abramov reaction is the related conversions of trialkyl to α-hydroxy phosphonates by the addition to carbonyl compounds. In terms of mechanism, the reaction involves attack of the nucleophilic phosphorus atom on the carbonyl carbon. It was named after the Russian chemist Vasilii Semenovich Abramov (1904–1968) in 1957.
The [2,3]-Wittig rearrangement is the transformation of an allylic ether into a homoallylic alcohol via a concerted, pericyclic process. Because the reaction is concerted, it exhibits a high degree of stereocontrol, and can be employed early in a synthetic route to establish stereochemistry. The Wittig rearrangement requires strongly basic conditions, however, as a carbanion intermediate is essential. [1,2]-Wittig rearrangement is a competitive process.
Oxidation with chromium(VI) complexes involves the conversion of alcohols to carbonyl compounds or more highly oxidized products through the action of molecular chromium(VI) oxides and salts. The principal reagents are Collins reagent, PDC, and PCC. These reagents represent improvements over inorganic chromium(VI) reagents such as Jones reagent.
Reductions with metal alkoxyaluminium hydrides are chemical reactions that involve either the net hydrogenation of an unsaturated compound or the replacement of a reducible functional group with hydrogen by metal alkoxyaluminium hydride reagents.
Benzylic activation and stereocontrol in tricarbonyl(arene)chromium complexes refers to the enhanced rates and stereoselectivities of reactions at the benzylic position of aromatic rings complexed to chromium(0) relative to uncomplexed arenes. Complexation of an aromatic ring to chromium stabilizes both anions and cations at the benzylic position and provides a steric blocking element for diastereoselective functionalization of the benzylic position. A large number of stereoselective methods for benzylic and homobenzylic functionalization have been developed based on this property.
Reactions of organocopper reagents involve species containing copper-carbon bonds acting as nucleophiles in the presence of organic electrophiles. Organocopper reagents are now commonly used in organic synthesis as mild, selective nucleophiles for substitution and conjugate addition reactions.
In organic chemistry, the Keck asymmetric allylation is a chemical reaction that involves the nucleophilic addition of an allyl group to an aldehyde. The catalyst is a chiral complex that contains titanium as a Lewis acid. The chirality of the catalyst induces a stereoselective addition, so the secondary alcohol of the product has a predictable absolute stereochemistry based on the choice of catalyst. This name reaction is named for Gary Keck.
In organic chemistry, carbonyl allylation describes methods for adding an allyl anion to an aldehyde or ketone to produce a homoallylic alcohol. The carbonyl allylation was first reported in 1876 by Alexander Zaitsev and employed an allylzinc reagent.

The Krische allylation involves the enantioselective iridium-catalyzed addition of an allyl group to an aldehyde or an alcohol, resulting in the formation of a secondary homoallylic alcohol. The mechanism of the Krische allylation involves primary alcohol dehydrogenation or, when using aldehyde reactants, hydrogen transfer from 2-propanol. Unlike other allylation methods, the Krische allylation avoids the use of preformed allyl metal reagents and enables the direct conversion of primary alcohols to secondary homoallylic alcohols.











