Inorganic chemistry deals with synthesis and behavior of inorganic and organometallic compounds. This field covers chemical compounds that are not carbon-based, which are the subjects of organic chemistry. The distinction between the two disciplines is far from absolute, as there is much overlap in the subdiscipline of organometallic chemistry. It has applications in every aspect of the chemical industry, including catalysis, materials science, pigments, surfactants, coatings, medications, fuels, and agriculture.

Redox is a type of chemical reaction in which the oxidation states of substrate change.
Ferredoxins are iron–sulfur proteins that mediate electron transfer in a range of metabolic reactions. The term "ferredoxin" was coined by D.C. Wharton of the DuPont Co. and applied to the "iron protein" first purified in 1962 by Mortenson, Valentine, and Carnahan from the anaerobic bacterium Clostridium pasteurianum.
Iron–sulfur proteins are proteins characterized by the presence of iron–sulfur clusters containing sulfide-linked di-, tri-, and tetrairon centers in variable oxidation states. Iron–sulfur clusters are found in a variety of metalloproteins, such as the ferredoxins, as well as NADH dehydrogenase, hydrogenases, coenzyme Q – cytochrome c reductase, succinate – coenzyme Q reductase and nitrogenase. Iron–sulfur clusters are best known for their role in the oxidation-reduction reactions of electron transport in mitochondria and chloroplasts. Both Complex I and Complex II of oxidative phosphorylation have multiple Fe–S clusters. They have many other functions including catalysis as illustrated by aconitase, generation of radicals as illustrated by SAM-dependent enzymes, and as sulfur donors in the biosynthesis of lipoic acid and biotin. Additionally, some Fe–S proteins regulate gene expression. Fe–S proteins are vulnerable to attack by biogenic nitric oxide, forming dinitrosyl iron complexes. In most Fe–S proteins, the terminal ligands on Fe are thiolate, but exceptions exist.
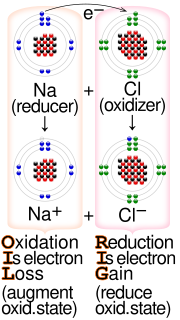
Electron transfer (ET) occurs when an electron relocates from an atom or molecule to another such chemical entity. ET is a mechanistic description of certain kinds of redox reactions involving transfer of electrons.
Inner sphere electron transfer or bonded electron transfer is a redox chemical reaction that proceeds via a covalent linkage—a strong electronic interaction—between the oxidant and the reductant reactants. In inner sphere electron transfer, a ligand bridges the two metal redox centers during the electron transfer event. Inner sphere reactions are inhibited by large ligands, which prevent the formation of the crucial bridged intermediate. Thus, inner sphere ET is rare in biological systems, where redox sites are often shielded by bulky proteins. Inner sphere ET is usually used to describe reactions involving transition metal complexes and most of this article is written from this perspective. However, redox centers can consist of organic groups rather than metal centers.
In theoretical chemistry, Marcus theory is a theory originally developed by Rudolph A. Marcus, starting in 1956, to explain the rates of electron transfer reactions – the rate at which an electron can move or jump from one chemical species (called the electron donor) to another (called the electron acceptor). It was originally formulated to address outer sphere electron transfer reactions, in which the two chemical species only change in their charge with an electron jumping (e.g. the oxidation of an ion like Fe2+/Fe3+), but do not undergo large structural changes. It was extended to include inner sphere electron transfer contributions, in which a change of distances or geometry in the solvation or coordination shells of the two chemical species is taken into account (the Fe-O distances in Fe(H2O)2+ and Fe(H2O)3+ are different).

A photosynthetic reaction center is a complex of several proteins, pigments and other co-factors that together execute the primary energy conversion reactions of photosynthesis. Molecular excitations, either originating directly from sunlight or transferred as excitation energy via light-harvesting antenna systems, give rise to electron transfer reactions along the path of a series of protein-bound co-factors. These co-factors are light-absorbing molecules (also named chromophores or pigments) such as chlorophyll and pheophytin, as well as quinones. The energy of the photon is used to excite an electron of a pigment. The free energy created is then used, via a chain of nearby electron acceptors, for a transfer of hydrogen atoms (as protons and electrons) from H2O or hydrogen sulfide towards carbon dioxide, eventually producing glucose. These electron transfer steps ultimately result in the conversion of the energy of photons to chemical energy.
Rubredoxins are a class of low-molecular-weight iron-containing proteins found in sulfur-metabolizing bacteria and archaea. Sometimes rubredoxins are classified as iron-sulfur proteins; however, in contrast to iron-sulfur proteins, rubredoxins do not contain inorganic sulfide. Like cytochromes, ferredoxins and Rieske proteins, rubredoxins participate in electron transfer in biological systems.

In coordination chemistry, the first coordination sphere refers to the array of molecules and ions directly attached to the central metal atom. The second coordination sphere consists of molecules and ions that attached in various ways to the first coordination sphere.

In enzymology, ferredoxin hydrogenase, also referred to as [Fe-Fe]hydrogenase, H2 oxidizing hydrogenase, H2 producing hydrogenase, bidirectional hydrogenase, hydrogenase (ferredoxin), hydrogenlyase, and uptake hydrogenase, is found in Clostridium pasteurianum, Clostridium acetobutylicum,Chlamydomonas reinhardtii, and other organisms. The systematic name of this enzyme is hydrogen:ferredoxin oxidoreductase
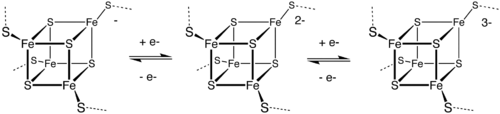
High potential iron-sulfur proteins (HIPIP) are a specific class of high-redox potential 4Fe-4S ferredoxins that functions in anaerobic electron transport and which occurs in photosynthetic bacteria and in Paracoccus denitrificans. The HiPIPs are small proteins which show significant variation in their sequences, their sizes, and in their oxidation- reduction potentials. As shown in the following schematic representation the iron-sulfur cluster is bound by four conserved cysteine residues.
[ 4Fe-4S cluster] | | | | xxxxxxxxxxxxxxxxxxxCxCxxxxxxxCxxxxxCxxxx
A Proton-coupled electron transfer (PCET) is a chemical reaction that involves the transfer of electrons and protons from one atom to another. The term was originally coined for single proton, single electron processes that are concerted, but the definition has relaxed to include many related processes. Reactions that involve the concerted shift of a single electron and a single proton are often called Concerted Proton-Electron Transfer or CPET.
Spin states when describing transition metal coordination complexes refers to the potential spin configurations of the central metal's d electrons. For several oxidation states, metals can adopt high-spin and low-spin configurations. The ambiguity only applies to first row metals, because second- and third-row metals are invariably low-spin. These configurations can be understood through the two major models used to describe coordination complexes; crystal field theory and ligand field theory.
In chemistry, metal aquo complexes are coordination compounds containing metal ions with only water as a ligand. These complexes are the predominant species in aqueous solutions of many metal salts, such as metal nitrates, sulfates, and perchlorates. They have the general stoichiometry [M(H2O)n]z+. Their behavior underpins many aspects of environmental, biological, and industrial chemistry. This article focuses on complexes where water is the only ligand, but of course many complexes are known to consist of a mix of aquo and other ligands.
[NiFe] hydrogenase is a type of hydrogenase, which is an oxidative enzyme that reversibly converts molecular hydrogen in prokaryotes including Bacteria and Archaea. The catalytic site on the enzyme provides simple hydrogen-metabolizing microorganisms a redox mechanism by which to store and utilize energy via the reaction shown in Figure 1. This is particularly essential for the anaerobic, sulfate-reducing bacteria of the genus Desulfovibrio as well as pathogenic organisms Escherichia coli and Helicobacter pylori. The mechanisms, maturation, and function of [NiFe] hydrogenases are actively being researched for applications to the hydrogen economy and as potential antibiotic targets.
Photoredox catalysis is a branch of photochemistry that uses single-electron transfer. Photoredox catalysts are generally drawn from three classes of materials: transition-metal complexes, organic dyes, and semiconductors. While organic photoredox catalysts were dominant throughout the 1990s and early 2000s, soluble transition-metal complexes are more commonly used today.
In electrochemistry, protein film voltammetry is a technique for examining the behavior of proteins immobilized on an electrode. The technique is applicable to proteins and enzymes that engage in electron transfer reactions and it is part of the methods available to study enzyme kinetics.

Transition metal complexes of nitrite describes families of coordination complexes containing one or more nitrite ligands. Although the synthetic derivatives are only of scholarly interest, metal-nitrite complexes occur in several enzymes that participate in the nitrogen cycle.
Transition metal complexes of 2,2'-bipyridine are coordination complexes containing one or more 2,2'-bipyridine ligands. Complexes have been described for all of the transition metals. Although few have any practical value, these complexes have been influential. 2,2'-Bipyridine is classified as a diimine ligand. Unlike the structures of pyridine complexes, the two rings in bipy are coplanar, which facilitates electron delocalization. As a consequence of this delocalization, bipy complexes often exhibit distinctive optical and redox properties.