
In organic chemistry, allenes are organic compounds in which one carbon atom has double bonds with each of its two adjacent carbon atoms. Allenes are classified as cumulated dienes. The parent compound of this class is propadiene, which is itself also called allene. An group of the structure R2C=C=CR− is called allenyl, where R is H or some alkyl group. Compounds with an allene-type structure but with more than three carbon atoms are members of a larger class of compounds called cumulenes with X=C=Y bonding.
Computational chemistry is a branch of chemistry that uses computer simulation to assist in solving chemical problems. It uses methods of theoretical chemistry, incorporated into computer programs, to calculate the structures and properties of molecules, groups of molecules, and solids. It is essential because, apart from relatively recent results concerning the hydrogen molecular ion, the quantum many-body problem cannot be solved analytically, much less in closed form. While computational results normally complement the information obtained by chemical experiments, it can in some cases predict hitherto unobserved chemical phenomena. It is widely used in the design of new drugs and materials.

A covalent bond is a chemical bond that involves the sharing of electrons to form electron pairs between atoms. These electron pairs are known as shared pairs or bonding pairs. The stable balance of attractive and repulsive forces between atoms, when they share electrons, is known as covalent bonding. For many molecules, the sharing of electrons allows each atom to attain the equivalent of a full valence shell, corresponding to a stable electronic configuration. In organic chemistry, covalent bonding is much more common than ionic bonding.
A molecule is a group of two or more atoms held together by attractive forces known as chemical bonds; depending on context, the term may or may not include ions which satisfy this criterion. In quantum physics, organic chemistry, and biochemistry, the distinction from ions is dropped and molecule is often used when referring to polyatomic ions.
Quantum chemistry, also called molecular quantum mechanics, is a branch of physical chemistry focused on the application of quantum mechanics to chemical systems, particularly towards the quantum-mechanical calculation of electronic contributions to physical and chemical properties of molecules, materials, and solutions at the atomic level. These calculations include systematically applied approximations intended to make calculations computationally feasible while still capturing as much information about important contributions to the computed wave functions as well as to observable properties such as structures, spectra, and thermodynamic properties. Quantum chemistry is also concerned with the computation of quantum effects on molecular dynamics and chemical kinetics.

Molecular modelling encompasses all methods, theoretical and computational, used to model or mimic the behaviour of molecules. The methods are used in the fields of computational chemistry, drug design, computational biology and materials science to study molecular systems ranging from small chemical systems to large biological molecules and material assemblies. The simplest calculations can be performed by hand, but inevitably computers are required to perform molecular modelling of any reasonably sized system. The common feature of molecular modelling methods is the atomistic level description of the molecular systems. This may include treating atoms as the smallest individual unit, or explicitly modelling protons and neutrons with its quarks, anti-quarks and gluons and electrons with its photons.
In chemistry, orbital hybridisation is the concept of mixing atomic orbitals to form new hybrid orbitals suitable for the pairing of electrons to form chemical bonds in valence bond theory. For example, in a carbon atom which forms four single bonds the valence-shell s orbital combines with three valence-shell p orbitals to form four equivalent sp3 mixtures in a tetrahedral arrangement around the carbon to bond to four different atoms. Hybrid orbitals are useful in the explanation of molecular geometry and atomic bonding properties and are symmetrically disposed in space. Usually hybrid orbitals are formed by mixing atomic orbitals of comparable energies.
In quantum chemistry, the quantum theory of atoms in molecules (QTAIM), sometimes referred to as atoms in molecules (AIM), is a model of molecular and condensed matter electronic systems in which the principal objects of molecular structure - atoms and bonds - are natural expressions of a system's observable electron density distribution function. An electron density distribution of a molecule is a probability distribution that describes the average manner in which the electronic charge is distributed throughout real space in the attractive field exerted by the nuclei. According to QTAIM, molecular structure is revealed by the stationary points of the electron density together with the gradient paths of the electron density that originate and terminate at these points.

In the context of chemistry and molecular modelling, a force field is a computational method that is used to estimate the forces between atoms within molecules and also between molecules. More precisely, the force field refers to the functional form and parameter sets used to calculate the potential energy of a system of atoms or coarse-grained particles in molecular mechanics, molecular dynamics, or Monte Carlo simulations. The parameters for a chosen energy function may be derived from experiments in physics and chemistry, calculations in quantum mechanics, or both. Force fields are interatomic potentials and utilize the same concept as force fields in classical physics, with the difference that the force field parameters in chemistry describe the energy landscape, from which the acting forces on every particle are derived as a gradient of the potential energy with respect to the particle coordinates.
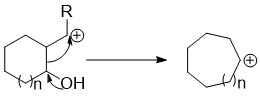
Ring expansion and ring contraction reactions expand or contract rings, usually in organic chemistry. The term usually refers to reactions involve making and breaking C-C bonds, Diverse mechanisms llead to these kinds of reactions.

Michael James Steuart Dewar was an American theoretical chemist.

Spartan is a molecular modelling and computational chemistry application from Wavefunction. It contains code for molecular mechanics, semi-empirical methods, ab initio models, density functional models, post-Hartree–Fock models, and thermochemical recipes including G3(MP2) and T1. Quantum chemistry calculations in Spartan are powered by Q-Chem.
Semi-empirical quantum chemistry methods are based on the Hartree–Fock formalism, but make many approximations and obtain some parameters from empirical data. They are very important in computational chemistry for treating large molecules where the full Hartree–Fock method without the approximations is too expensive. The use of empirical parameters appears to allow some inclusion of electron correlation effects into the methods.
Ab initio quantum chemistry methods are computational chemistry methods based on quantum chemistry. The term ab initio was first used in quantum chemistry by Robert Parr and coworkers, including David Craig in a semiempirical study on the excited states of benzene. The background is described by Parr. Ab initio means "from first principles" or "from the beginning", implying that the only inputs into an ab initio calculation are physical constants. Ab initio quantum chemistry methods attempt to solve the electronic Schrödinger equation given the positions of the nuclei and the number of electrons in order to yield useful information such as electron densities, energies and other properties of the system. The ability to run these calculations has enabled theoretical chemists to solve a range of problems and their importance is highlighted by the awarding of the Nobel prize to John Pople and Walter Kohn.
Car–Parrinello molecular dynamics or CPMD refers to either a method used in molecular dynamics or the computational chemistry software package used to implement this method.
Localized molecular orbitals are molecular orbitals which are concentrated in a limited spatial region of a molecule, such as a specific bond or lone pair on a specific atom. They can be used to relate molecular orbital calculations to simple bonding theories, and also to speed up post-Hartree–Fock electronic structure calculations by taking advantage of the local nature of electron correlation. Localized orbitals in systems with periodic boundary conditions are known as Wannier functions.
Quantum chemistry composite methods are computational chemistry methods that aim for high accuracy by combining the results of several calculations. They combine methods with a high level of theory and a small basis set with methods that employ lower levels of theory with larger basis sets. They are commonly used to calculate thermodynamic quantities such as enthalpies of formation, atomization energies, ionization energies and electron affinities. They aim for chemical accuracy which is usually defined as within 1 kcal/mol of the experimental value. The first systematic model chemistry of this type with broad applicability was called Gaussian-1 (G1) introduced by John Pople. This was quickly replaced by the Gaussian-2 (G2) which has been used extensively. The Gaussian-3 (G3) was introduced later.
The Stieglitz rearrangement is a rearrangement reaction in organic chemistry which is named after the American chemist Julius Stieglitz (1867–1937) and was first investigated by him and Paul Nicholas Leech in 1913. It describes the 1,2-rearrangement of trityl amine derivatives to triaryl imines. It is comparable to a Beckmann rearrangement which also involves a substitution at a nitrogen atom through a carbon to nitrogen shift. As an example, triaryl hydroxylamines can undergo a Stieglitz rearrangement by dehydration and the shift of a phenyl group after activation with phosphorus pentachloride to yield the respective triaryl imine, a Schiff base.
In the context of chemistry and molecular modelling, the Interface force field (IFF) is a force field for classical molecular simulations of atoms, molecules, and assemblies up to the large nanometer scale, covering compounds from across the periodic table. It employs a consistent classical Hamiltonian energy function for metals, oxides, and organic compounds, linking biomolecular and materials simulation platforms into a single platform. The reliability is often higher than that of density functional theory calculations at more than a million times lower computational cost. IFF includes a physical-chemical interpretation for all parameters as well as a surface model database that covers different cleavage planes and surface chemistry of included compounds. The Interface Force Field is compatible with force fields for the simulation of primarily organic compounds and can be used with common molecular dynamics and Monte Carlo codes. Structures and energies of included chemical elements and compounds are rigorously validated and property predictions are up to a factor of 100 more accurate relative to earlier models.

Imre Gyula Csizmadia was a Canadian Hungarian chemist, university professor, external member of the Hungarian Academy of Sciences.


