
An alpha helix is a sequence of amino acids in a protein that are twisted into a coil.
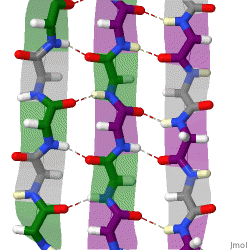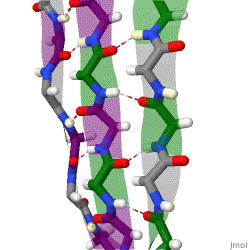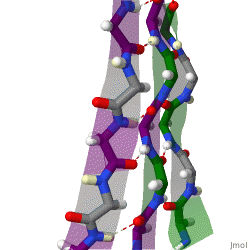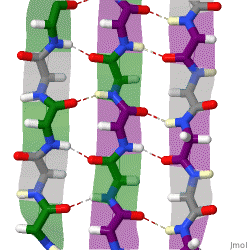
The beta sheet, (β-sheet) is a common motif of the regular protein secondary structure. Beta sheets consist of beta strands (β-strands) connected laterally by at least two or three backbone hydrogen bonds, forming a generally twisted, pleated sheet. A β-strand is a stretch of polypeptide chain typically 3 to 10 amino acids long with backbone in an extended conformation. The supramolecular association of β-sheets has been implicated in the formation of the fibrils and protein aggregates observed in amyloidosis, Alzheimer's disease and other proteinopathies.

Protein secondary structure is the local spatial conformation of the polypeptide backbone excluding the side chains. The two most common secondary structural elements are alpha helices and beta sheets, though beta turns and omega loops occur as well. Secondary structure elements typically spontaneously form as an intermediate before the protein folds into its three dimensional tertiary structure.

Protein tertiary structure is the three dimensional shape of a protein. The tertiary structure will have a single polypeptide chain "backbone" with one or more protein secondary structures, the protein domains. Amino acid side chains may interact and bond in a number of ways. The interactions and bonds of side chains within a particular protein determine its tertiary structure. The protein tertiary structure is defined by its atomic coordinates. These coordinates may refer either to a protein domain or to the entire tertiary structure. A number of tertiary structures may fold into a quaternary structure.

A transmembrane protein (TP) is a type of integral membrane protein that spans the entirety of the cell membrane. Many transmembrane proteins function as gateways to permit the transport of specific substances across the membrane. They frequently undergo significant conformational changes to move a substance through the membrane. They are usually highly hydrophobic and aggregate and precipitate in water. They require detergents or nonpolar solvents for extraction, although some of them (beta-barrels) can be also extracted using denaturing agents.

In biology and biochemistry, the active site is the region of an enzyme where substrate molecules bind and undergo a chemical reaction. The active site consists of amino acid residues that form temporary bonds with the substrate, the binding site, and residues that catalyse a reaction of that substrate, the catalytic site. Although the active site occupies only ~10–20% of the volume of an enzyme, it is the most important part as it directly catalyzes the chemical reaction. It usually consists of three to four amino acids, while other amino acids within the protein are required to maintain the tertiary structure of the enzymes.

Protein structure prediction is the inference of the three-dimensional structure of a protein from its amino acid sequence—that is, the prediction of its secondary and tertiary structure from primary structure. Structure prediction is different from the inverse problem of protein design. Protein structure prediction is one of the most important goals pursued by computational biology; and it is important in medicine and biotechnology.

Structural alignment attempts to establish homology between two or more polymer structures based on their shape and three-dimensional conformation. This process is usually applied to protein tertiary structures but can also be used for large RNA molecules. In contrast to simple structural superposition, where at least some equivalent residues of the two structures are known, structural alignment requires no a priori knowledge of equivalent positions. Structural alignment is a valuable tool for the comparison of proteins with low sequence similarity, where evolutionary relationships between proteins cannot be easily detected by standard sequence alignment techniques. Structural alignment can therefore be used to imply evolutionary relationships between proteins that share very little common sequence. However, caution should be used in using the results as evidence for shared evolutionary ancestry because of the possible confounding effects of convergent evolution by which multiple unrelated amino acid sequences converge on a common tertiary structure.

Structural bioinformatics is the branch of bioinformatics that is related to the analysis and prediction of the three-dimensional structure of biological macromolecules such as proteins, RNA, and DNA. It deals with generalizations about macromolecular 3D structures such as comparisons of overall folds and local motifs, principles of molecular folding, evolution, binding interactions, and structure/function relationships, working both from experimentally solved structures and from computational models. The term structural has the same meaning as in structural biology, and structural bioinformatics can be seen as a part of computational structural biology. The main objective of structural bioinformatics is the creation of new methods of analysing and manipulating biological macromolecular data in order to solve problems in biology and generate new knowledge.

Protein structure is the three-dimensional arrangement of atoms in an amino acid-chain molecule. Proteins are polymers – specifically polypeptides – formed from sequences of amino acids, which are the monomers of the polymer. A single amino acid monomer may also be called a residue, which indicates a repeating unit of a polymer. Proteins form by amino acids undergoing condensation reactions, in which the amino acids lose one water molecule per reaction in order to attach to one another with a peptide bond. By convention, a chain under 30 amino acids is often identified as a peptide, rather than a protein. To be able to perform their biological function, proteins fold into one or more specific spatial conformations driven by a number of non-covalent interactions, such as hydrogen bonding, ionic interactions, Van der Waals forces, and hydrophobic packing. To understand the functions of proteins at a molecular level, it is often necessary to determine their three-dimensional structure. This is the topic of the scientific field of structural biology, which employs techniques such as X-ray crystallography, NMR spectroscopy, cryo-electron microscopy (cryo-EM) and dual polarisation interferometry, to determine the structure of proteins.

Protein–protein interactions (PPIs) are physical contacts of high specificity established between two or more protein molecules as a result of biochemical events steered by interactions that include electrostatic forces, hydrogen bonding and the hydrophobic effect. Many are physical contacts with molecular associations between chains that occur in a cell or in a living organism in a specific biomolecular context.

In protein structures, a beta barrel is a beta sheet composed of tandem repeats that twists and coils to form a closed toroidal structure in which the first strand is bonded to the last strand. Beta-strands in many beta-barrels are arranged in an antiparallel fashion. Beta barrel structures are named for resemblance to the barrels used to contain liquids. Most of them are water-soluble proteins and frequently bind hydrophobic ligands in the barrel center, as in lipocalins. Others span cell membranes and are commonly found in porins. Porin-like barrel structures are encoded by as many as 2–3% of the genes in Gram-negative bacteria. It has been shown that more than 600 proteins with various function contain the beta barrel structure.
Biomolecular structure is the intricate folded, three-dimensional shape that is formed by a molecule of protein, DNA, or RNA, and that is important to its function. The structure of these molecules may be considered at any of several length scales ranging from the level of individual atoms to the relationships among entire protein subunits. This useful distinction among scales is often expressed as a decomposition of molecular structure into four levels: primary, secondary, tertiary, and quaternary. The scaffold for this multiscale organization of the molecule arises at the secondary level, where the fundamental structural elements are the molecule's various hydrogen bonds. This leads to several recognizable domains of protein structure and nucleic acid structure, including such secondary-structure features as alpha helixes and beta sheets for proteins, and hairpin loops, bulges, and internal loops for nucleic acids. The terms primary, secondary, tertiary, and quaternary structure were introduced by Kaj Ulrik Linderstrøm-Lang in his 1951 Lane Medical Lectures at Stanford University.
A turn is an element of secondary structure in proteins where the polypeptide chain reverses its overall direction.

The beta hairpin is a simple protein structural motif involving two beta strands that look like a hairpin. The motif consists of two strands that are adjacent in primary structure, oriented in an antiparallel direction, and linked by a short loop of two to five amino acids. Beta hairpins can occur in isolation or as part of a series of hydrogen bonded strands that collectively comprise a beta sheet.
Computational Resources for Drug Discovery (CRDD) is one of the important silico modules of Open Source for Drug Discovery (OSDD). The CRDD web portal provides computer resources related to drug discovery on a single platform. It provides computational resources for researchers in computer-aided drug design, a discussion forum, and resources to maintain a Wikipedia related to drug discovery, predict inhibitors, and predict the ADME-Tox property of molecules. One of the major objectives of CRDD is to promote open source software in the field of chemoinformatics and pharmacoinformatics.

Schellman loops are commonly occurring structural features of proteins and polypeptides. Each has six amino acid residues with two specific inter-mainchain hydrogen bonds and a characteristic main chain dihedral angle conformation. The CO group of residue i is hydrogen-bonded to the NH of residue i+5, and the CO group of residue i+1 is hydrogen-bonded to the NH of residue i+4. Residues i+1, i+2, and i+3 have negative φ (phi) angle values and the phi value of residue i+4 is positive. Schellman loops incorporate a three amino acid residue RL nest, in which three mainchain NH groups form a concavity for hydrogen bonding to carbonyl oxygens. About 2.5% of amino acids in proteins belong to Schellman loops. Two websites are available for examining small motifs in proteins, Motivated Proteins: ; or PDBeMotif:.
PSI-blast based secondary structure PREDiction (PSIPRED) is a method used to investigate protein structure. It uses artificial neural network machine learning methods in its algorithm. It is a server-side program, featuring a website serving as a front-end interface, which can predict a protein's secondary structure from the primary sequence.
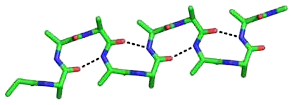
The beta bend ribbon, or beta-bend ribbon, is a structural feature in polypeptides and proteins. The shortest possible has six amino acid residues arranged as two overlapping hydrogen bonded beta turns in which the carbonyl group of residue i is hydrogen-bonded to the NH of residue i+3 while the carbonyl group of residue i+2 is hydrogen-bonded to the NH of residue i+5. In longer ribbons, this bonding is continued in peptides of 8, 10, etc., amino acid residues. A beta bend ribbon can be regarded as an aberrant 310 helix (3/10-helix) that has lost some of its hydrogen bonds. Two websites are available to facilitate finding and examining these features in proteins: Motivated Proteins; and PDBeMotif.
Amide Rings are small motifs in proteins and polypeptides. They consist of 9-atom or 11-atom rings formed by two CO...HN hydrogen bonds between a side chain amide group and the main chain atoms of a short polypeptide. They are observed with glutamine or asparagine side chains within proteins and polypeptides. Structurally similar rings occur in the binding of purine, pyrimidine and nicotinamide bases to the main chain atoms of proteins. About 4% of asparagines and glutamines form amide rings; in databases of protein domain structures, one is present, on average, every other protein.







