Related Research Articles

The polymerase chain reaction (PCR) is a method widely used to make millions to billions of copies of a specific DNA sample rapidly, allowing scientists to amplify a very small sample of DNA sufficiently to enable detailed study. PCR was invented in 1983 by American biochemist Kary Mullis at Cetus Corporation. Mullis and biochemist Michael Smith, who had developed other essential ways of manipulating DNA, were jointly awarded the Nobel Prize in Chemistry in 1993.

Reverse transcription polymerase chain reaction (RT-PCR) is a laboratory technique combining reverse transcription of RNA into DNA and amplification of specific DNA targets using polymerase chain reaction (PCR). It is primarily used to measure the amount of a specific RNA. This is achieved by monitoring the amplification reaction using fluorescence, a technique called real-time PCR or quantitative PCR (qPCR). Confusion can arise because some authors use the acronym RT-PCR to denote real-time PCR. In this article, RT-PCR will denote Reverse Transcription PCR. Combined RT-PCR and qPCR are routinely used for analysis of gene expression and quantification of viral RNA in research and clinical settings.

The thermal cycler is a laboratory apparatus most commonly used to amplify segments of DNA via the polymerase chain reaction (PCR). Thermal cyclers may also be used in laboratories to facilitate other temperature-sensitive reactions, including restriction enzyme digestion or rapid diagnostics. The device has a thermal block with holes where tubes holding the reaction mixtures can be inserted. The cycler then raises and lowers the temperature of the block in discrete, pre-programmed steps.
Cycling probe technology (CPT) is a molecular biological technique for detecting specific DNA sequences. CPT operates under isothermal conditions. In some applications, CPT offers an alternative to PCR. However, unlike PCR, CPT does not generate multiple copies of the target DNA itself, and the amplification of the signal is linear, in contrast to the exponential amplification of the target DNA in PCR. CPT uses a sequence specific chimeric probe which hybridizes to a complementary target DNA sequence and becomes a substrate for RNase H. Cleavage occurs at the RNA internucleotide linkages and results in dissociation of the probe from the target, thereby making it available for the next probe molecule. Integrated electrokinetic systems have been developed for use in CPT.
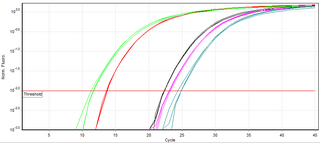
A real-time polymerase chain reaction is a laboratory technique of molecular biology based on the polymerase chain reaction (PCR). It monitors the amplification of a targeted DNA molecule during the PCR, not at its end, as in conventional PCR. Real-time PCR can be used quantitatively and semi-quantitatively.
TaqMan probes are hydrolysis probes that are designed to increase the specificity of quantitative PCR. The method was first reported in 1991 by researcher Kary Mullis at Cetus Corporation, and the technology was subsequently developed by Hoffmann-La Roche for diagnostic assays and by Applied Biosystems for research applications.
SNP genotyping is the measurement of genetic variations of single nucleotide polymorphisms (SNPs) between members of a species. It is a form of genotyping, which is the measurement of more general genetic variation. SNPs are one of the most common types of genetic variation. An SNP is a single base pair mutation at a specific locus, usually consisting of two alleles. SNPs are found to be involved in the etiology of many human diseases and are becoming of particular interest in pharmacogenetics. Because SNPs are conserved during evolution, they have been proposed as markers for use in quantitative trait loci (QTL) analysis and in association studies in place of microsatellites. The use of SNPs is being extended in the HapMap project, which aims to provide the minimal set of SNPs needed to genotype the human genome. SNPs can also provide a genetic fingerprint for use in identity testing. The increase of interest in SNPs has been reflected by the furious development of a diverse range of SNP genotyping methods.

Bisulfitesequencing (also known as bisulphite sequencing) is the use of bisulfite treatment of DNA before routine sequencing to determine the pattern of methylation. DNA methylation was the first discovered epigenetic mark, and remains the most studied. In animals it predominantly involves the addition of a methyl group to the carbon-5 position of cytosine residues of the dinucleotide CpG, and is implicated in repression of transcriptional activity.
Digital polymerase chain reaction is a biotechnological refinement of conventional polymerase chain reaction methods that can be used to directly quantify and clonally amplify nucleic acids strands including DNA, cDNA, or RNA. The key difference between dPCR and traditional PCR lies in the method of measuring nucleic acids amounts, with the former being a more precise method than PCR, though also more prone to error in the hands of inexperienced users. A "digital" measurement quantitatively and discretely measures a certain variable, whereas an “analog” measurement extrapolates certain measurements based on measured patterns. PCR carries out one reaction per single sample. dPCR also carries out a single reaction within a sample, however the sample is separated into a large number of partitions and the reaction is carried out in each partition individually. This separation allows a more reliable collection and sensitive measurement of nucleic acid amounts. The method has been demonstrated as useful for studying variations in gene sequences — such as copy number variants and point mutations — and it is routinely used for clonal amplification of samples for next-generation sequencing.
Loop-mediated isothermal amplification (LAMP) is a single-tube technique for the amplification of DNA and a low-cost alternative to detect certain diseases that was invented in 2000 at the University of Tokyo. Reverse transcription loop-mediated isothermal amplification (RT-LAMP) combines LAMP with a reverse transcription step to allow the detection of RNA.
Melting curve analysis is an assessment of the dissociation characteristics of double-stranded DNA during heating. As the temperature is raised, the double strand begins to dissociate leading to a rise in the absorbance intensity, hyperchromicity. The temperature at which 50% of DNA is denatured is known as the melting temperature. Measurement of melting temperature can help us predict species by just studying the melting temperature. This is because every organism has a specific melting curve.
The versatility of polymerase chain reaction (PCR) has led to modifications of the basic protocol being used in a large number of variant techniques designed for various purposes. This article summarizes many of the most common variations currently or formerly used in molecular biology laboratories; familiarity with the fundamental premise by which PCR works and corresponding terms and concepts is necessary for understanding these variant techniques.
Nucleic acid methods are the techniques used to study nucleic acids: DNA and RNA.
High Resolution Melt (HRM) analysis is a powerful technique in molecular biology for the detection of mutations, polymorphisms and epigenetic differences in double-stranded DNA samples. It was discovered and developed by Idaho Technology and the University of Utah. It has advantages over other genotyping technologies, namely:
A primer dimer (PD) is a potential by-product in the polymerase chain reaction (PCR), a common biotechnological method. As its name implies, a PD consists of two primer molecules that have attached (hybridized) to each other because of strings of complementary bases in the primers. As a result, the DNA polymerase amplifies the PD, leading to competition for PCR reagents, thus potentially inhibiting amplification of the DNA sequence targeted for PCR amplification. In quantitative PCR, PDs may interfere with accurate quantification.
COLD-PCR is a modified polymerase chain reaction (PCR) protocol that enriches variant alleles from a mixture of wildtype and mutation-containing DNA. The ability to preferentially amplify and identify minority alleles and low-level somatic DNA mutations in the presence of excess wildtype alleles is useful for the detection of mutations. Detection of mutations is important in the case of early cancer detection from tissue biopsies and body fluids such as blood plasma or serum, assessment of residual disease after surgery or chemotherapy, disease staging and molecular profiling for prognosis or tailoring therapy to individual patients, and monitoring of therapy outcome and cancer remission or relapse. Common PCR will amplify both the major (wildtype) and minor (mutant) alleles with the same efficiency, occluding the ability to easily detect the presence of low-level mutations. The capacity to detect a mutation in a mixture of variant/wildtype DNA is valuable because this mixture of variant DNAs can occur when provided with a heterogeneous sample – as is often the case with cancer biopsies. Currently, traditional PCR is used in tandem with a number of different downstream assays for genotyping or the detection of somatic mutations. These can include the use of amplified DNA for RFLP analysis, MALDI-TOF genotyping, or direct sequencing for detection of mutations by Sanger sequencing or pyrosequencing. Replacing traditional PCR with COLD-PCR for these downstream assays will increase the reliability in detecting mutations from mixed samples, including tumors and body fluids.
A thermal shift assay (TSA) measures changes in the thermal denaturation temperature and hence stability of a protein under varying conditions such as variations in drug concentration, buffer pH or ionic strength, redox potential, or sequence mutation. The most common method for measuring protein thermal shifts is differential scanning fluorimetry (DSF) or thermofluor, which utilizes specialized fluorogenic dyes.
Stephen Andrew Bustin is a British scientist, former professor of molecular sciences at Queen Mary University of London from 2004 to 2012, as well as visiting professor at Middlesex University, beginning in 2006. In 2012 he was appointed Professor of Allied Health and Medicine at Anglia Ruskin University. He is known for his research into polymerase chain reaction, and has written a book on the topic, entitled A-Z of Quantitative PCR. This book has been called "the bible of qPCR."
Recombinase polymerase amplification (RPA) is a single tube, isothermal alternative to the polymerase chain reaction (PCR). By adding a reverse transcriptase enzyme to an RPA reaction it can detect RNA as well as DNA, without the need for a separate step to produce cDNA,. Because it is isothermal, RPA can use much simpler equipment than PCR, which requires a thermal cycler. Operating best at temperatures of 37–42 °C and still working, albeit more slowly, at room temperature means RPA reactions can in theory be run quickly simply by holding a tube. This makes RPA an excellent candidate for developing low-cost, rapid, point-of-care molecular tests. An international quality assessment of molecular detection of Rift Valley fever virus performed as well as the best RT-PCR tests, detecting less concentrated samples missed by some PCR tests and an RT-LAMP test. RPA was developed and launched by TwistDx Ltd., a biotechnology company based in Cambridge, UK.

Reverse transcription loop-mediated isothermal amplification (RT-LAMP) is a one step nucleic acid amplification method to multiply specific sequences of RNA. It is used to diagnose infectious disease caused by RNA viruses.
References
- ↑ Also sometimes called "real-time PCR instrument".
- ↑ Higuchi, R.; Dollinger, G.; Walsh, P.S.; Griffith, R. (1992), "Simultaneous amplification and detection of specific DNA sequences", Bio/Technology, 10 (4): 413–7, doi:10.1038/nbt0492-413, PMID 1368485, S2CID 1684150
- 1 2 Logan, J.; Edwards, K. (January 2009). "Chapter 2 An Overview of PCR Platforms". In Saunders, N. (ed.). Real-Time PCR: Current Technology and Applications. Caister Academic Press. ISBN 978-1-904455-39-4.
- ↑ Open qPCR open source Real-Time PCR machine
- ↑ Ma, H.; Shieh, K.; Chen, G.; Chen, X.; Chuang, M. (2006), "Application of Real-time Polymerase Chain Reaction (RT-PCR)", The Journal of American Science, 2 (3): 1–15
- ↑ Raghavan, V.; Whitney, S.; Ebmeier, R.; Padhye, N.; Nelson, M. Viljoen; Gogos, G. (2006), "Thermal analysis of the vortex tube based thermocycler for fast DNA amplification: Experimental and two-dimensional numerical results", Review of Scientific Instruments, 77 (9): 094301–094301–9, Bibcode:2006RScI...77i4301R, doi:10.1063/1.2338283
- ↑ Wittwer, C.T.; Garling, D.J. (1991), "Rapid cycle DNA amplification: time and temperature optimization", BioTechniques, 10 (1): 76–83, PMID 2003928
- ↑ Kim, Y.H.; Yang, I.; Bae, Y.S.; Park, S.R. (2008), "Performance evaluation of thermal cyclers for PCR in a rapid cycling condition", BioTechniques, 44 (4): 495–6, doi: 10.2144/000112705 , PMID 18476814
- ↑ Schoder, D.; Schmalwieser, A.; Schauberger, G.; Kuhn, M.; Hoorfar, J.; Wagner, M. (2003), "Physical characteristics of six new thermocyclers", Clin. Chem., 49 (6): 960–3, doi:10.1373/49.6.960, PMID 12765996
- 1 2 Lee, D. -S. (2010). "Real-time PCR Machine System Modeling and a Systematic Approach for the Robust Design of a Real-time PCR-on-a-Chip System". Sensors. 10 (1): 697–718. Bibcode:2010Senso..10..697L. doi: 10.3390/s100100697 . PMC 3270864 . PMID 22315563.
- ↑ Schoder, D.; Schmalwieser, A.; Schauberger, G.; Hoorfar, J.; Kuhn, M.; Wagner, M. (2005), "Novel approach for assessing performance of PCR cyclers used for diagnostic testing", J Clin Microbiol, 43 (6): 2724–8, doi:10.1128/jcm.43.6.2724-2728.2005, PMC 1151936 , PMID 15956389
- ↑ Hilscher, C.; Vahrson, W.; Dittmer, D.P. (2005), "Faster quantitative real-time PCR protocols may lose sensitivity and show increased variability", Nucleic Acids Res., 33 (21): e182, doi:10.1093/nar/gni181, PMC 1297710 , PMID 16314296
- ↑ Herrmann, M.; Durtschi, J.; Wittwer, C.; Voelkerding, K. (2007). "Expanded instrument comparison of amplicon DNA melting analysis for mutation scanning and genotyping". Clinical Chemistry. 53 (8): 1544–1548. doi: 10.1373/clinchem.2007.088120 . PMID 17556647.
- 1 2 Gundry, C.; Vandersteen, J.; Reed, G.; Pryor, R.; Chen, J.; Wittwer, C. (2003). "Amplicon melting analysis with labeled primers: a closed-tube method for differentiating homozygotes and heterozygotes". Clinical Chemistry. 49 (3): 396–406. doi: 10.1373/49.3.396 . PMID 12600951.
- ↑ Köppel, R.; Zimmerli, F.; Breitenmoser, A. (2009), "Heptaplex real-time PCR for the identification and quantification of DNA from beef, pork, chicken, turkey, horse meat, sheep (mutton) and goat", European Food Research and Technology, 230: 125–33, doi:10.1007/s00217-009-1154-5, S2CID 96340566
- ↑ Bahrdt, C.; Krech, A.; Wurz, A.; Wulff, D. (2010). "Validation of a newly developed hexaplex real-time PCR assay for screening for presence of GMOs in food, feed and seed". Analytical and Bioanalytical Chemistry. 396 (6): 2103–2112. doi:10.1007/s00216-009-3380-x. PMID 20101506. S2CID 22657985.