Related Research Articles
Sharpless asymmetric dihydroxylation is the chemical reaction of an alkene with osmium tetroxide in the presence of a chiral quinine ligand to form a vicinal diol. The reaction has been applied to alkenes of virtually every substitution, often high enantioselectivities are realized, with the chiral outcome controlled by the choice of dihydroquinidine (DHQD) vs dihydroquinine (DHQ) as the ligand. Asymmetric dihydroxylation reactions are also highly site selective, providing products derived from reaction of the most electron-rich double bond in the substrate.
In chemistry, dehydrogenation is a chemical reaction that involves the removal of hydrogen, usually from an organic molecule. It is the reverse of hydrogenation. Dehydrogenation is important, both as a useful reaction and a serious problem. At its simplest, it is useful way of converting alkanes, which are relatively inert and thus low-valued, to olefins, which are reactive and thus more valuable. Alkenes are precursors to aldehydes, alcohols, polymers, and aromatics. As a problematic reaction, the fouling and inactivation of many catalysts arises via coking, which is the dehydrogenative polymerization of organic substrates.
The Stille reaction is a chemical reaction widely used in organic synthesis. The reaction involves the coupling of two organic groups, one of which is carried as an organotin compound (also known as organostannanes). A variety of organic electrophiles provide the other coupling partner. The Stille reaction is one of many palladium-catalyzed coupling reactions.
The Suzuki reaction is an organic reaction, classified as a cross-coupling reaction, where the coupling partners are a boronic acid and an organohalide and the catalyst is a palladium(0) complex. It was first published in 1979 by Akira Suzuki, and he shared the 2010 Nobel Prize in Chemistry with Richard F. Heck and Ei-ichi Negishi for their contribution to the discovery and development of palladium-catalyzed cross-couplings in organic synthesis. This reaction is also known as the Suzuki–Miyaura reaction or simply as the Suzuki coupling. It is widely used to synthesize polyolefins, styrenes, and substituted biphenyls. Several reviews have been published describing advancements and the development of the Suzuki reaction. The general scheme for the Suzuki reaction is shown below, where a carbon-carbon single bond is formed by coupling a halide (R1-X) with an organoboron species (R2-BY2) using a palladium catalyst and a base. The organoboron species is usually synthesized by hydroboration or carboboration, allowing for rapid generation of molecular complexity.
The Sonogashira reaction is a cross-coupling reaction used in organic synthesis to form carbon–carbon bonds. It employs a palladium catalyst as well as copper co-catalyst to form a carbon–carbon bond between a terminal alkyne and an aryl or vinyl halide.
Organopalladium chemistry is a branch of organometallic chemistry that deals with organic palladium compounds and their reactions. Palladium is often used as a catalyst in the reduction of alkenes and alkynes with hydrogen. This process involves the formation of a palladium-carbon covalent bond. Palladium is also prominent in carbon-carbon coupling reactions, as demonstrated in tandem reactions.

In organic chemistry, a sulfoxide, also called a sulfoxide, is an organosulfur compound containing a sulfinyl functional group attached to two carbon atoms. It is a polar functional group. Sulfoxides are oxidized derivatives of sulfides. Examples of important sulfoxides are alliin, a precursor to the compound that gives freshly crushed garlic its aroma, and dimethyl sulfoxide (DMSO), a common solvent.
Dihydroxylation is the process by which an alkene is converted into a vicinal diol. Although there are many routes to accomplish this oxidation, the most common and direct processes use a high-oxidation-state transition metal. The metal is often used as a catalyst, with some other stoichiometric oxidant present. In addition, other transition metals and non-transition metal methods have been developed and used to catalyze the reaction.
Ruthenium tetroxide is the inorganic compound with the formula RuO4. It is a yellow volatile solid that melts near room temperature. It has the odor of ozone. Samples are typically black due to impurities. The analogous OsO4 is more widely used and better known. It is also the anhydride of hyperruthenic acid (H2RuO5). One of the few solvents in which RuO4 forms stable solutions is CCl4.

A Grignard reagent or Grignard compound is a chemical compound with the general formula R−Mg−X, where X is a halogen and R is an organic group, normally an alkyl or aryl. Two typical examples are methylmagnesium chloride Cl−Mg−CH3 and phenylmagnesium bromide (C6H5)−Mg−Br. They are a subclass of the organomagnesium compounds.

Vanadium compounds are compounds formed by the element vanadium (V). The chemistry of vanadium is noteworthy for the accessibility of the four adjacent oxidation states 2–5, whereas the chemistry of the other group 5 elements, niobium and tantalum, are somewhat more limited to the +5 oxidation state. In aqueous solution, vanadium forms metal aquo complexes of which the colours are lilac [V(H2O)6]2+, green [V(H2O)6]3+, blue [VO(H2O)5]2+, yellow-orange oxides [VO(H2O)5]3+, the formula for which depends on pH. Vanadium(II) compounds are reducing agents, and vanadium(V) compounds are oxidizing agents. Vanadium(IV) compounds often exist as vanadyl derivatives, which contain the VO2+ center.
The Milas hydroxylation is an organic reaction converting an alkene to a vicinal diol, and was developed by Nicholas A. Milas in the 1930s. The cis-diol is formed by reaction of alkenes with hydrogen peroxide and either ultraviolet light or a catalytic osmium tetroxide, vanadium pentoxide, or chromium trioxide.
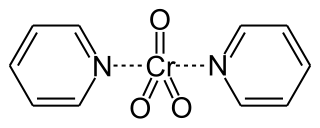
Collins reagent is the complex of chromium(VI) oxide with pyridine in dichloromethane. This metal-pyridine complex, a red solid, is used to oxidize primary alcohols to the corresponding aldehydes and secondary alcohols to the corresponding ketones. This complex is a hygroscopic orange solid.
In organic chemistry, the Kumada coupling is a type of cross coupling reaction, useful for generating carbon–carbon bonds by the reaction of a Grignard reagent and an organic halide. The procedure uses transition metal catalysts, typically nickel or palladium, to couple a combination of two alkyl, aryl or vinyl groups. The groups of Robert Corriu and Makoto Kumada reported the reaction independently in 1972.

The Shilov system is a classic example of catalytic C-H bond activation and oxidation which preferentially activates stronger C-H bonds over weaker C-H bonds for an overall partial oxidation.
Organovanadium chemistry is the chemistry of organometallic compounds containing a carbon (C) to vanadium (V) chemical bond. Organovanadium compounds find only minor use as reagents in organic synthesis but are significant for polymer chemistry as catalysts.
Alcohol oxidation is a class of organic reactions in which the alcohol functional group is converted into another functional group in which carbon carries a higher oxidation state.

Oxoammonium-catalyzed oxidation reactions involve the conversion of organic substrates to more highly oxidized materials through the action of an N-oxoammonium species. Nitroxides may also be used in catalytic amounts in the presence of a stoichiometric amount of a terminal oxidant. Nitroxide radical species used are either 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO) or derivatives thereof.

(2,2,6,6-Tetramethylpiperidin-1-yl)oxyl or (2,2,6,6-tetramethylpiperidin-1-yl)oxidanyl, commonly known as TEMPO, is a chemical compound with the formula (CH2)3(CMe2)2NO. This heterocyclic compound is a red-orange, sublimable solid. As a stable aminoxyl radical, it has applications in chemistry and biochemistry. TEMPO is used as a radical marker, as a structural probe for biological systems in conjunction with electron spin resonance spectroscopy, as a reagent in organic synthesis, and as a mediator in controlled radical polymerization.
Sulfonium-based oxidations of alcohols to aldehydes summarizes a group of organic reactions that transform a primary alcohol to the corresponding aldehyde (and a secondary alcohol to the corresponding ketone). Selective oxidation of alcohols to aldehydes requires circumventing over-oxidation to the carboxylic acid. One popular approach are methods that proceed through intermediate alkoxysulfonium species (RO−SMe+
2X-, e.g. compound 6) as detailed here. Since most of these methods employ dimethylsulfoxide (DMSO) as oxidant and generate dimethylsulfide, these are often colloquially summarized as DMSO-oxidations. Conceptually, generating an aldehyde and dimethylsulfide from an alcohol and DMSO requires a dehydrating agent for removal of H2O, ideally an electrophile simultaneously activating DMSO. In contrast, methods generating the sulfonium intermediate from dimethylsulfide do not require a dehydrating agent. Closely related are oxidations mediated by dimethyl selenoxide and by dimethyl selenide.
References
- ↑ Timothy J. Donohoe; Katherine M. P. Wheelhouse (née Gosby); Peter J. Lindsay‐Scott; Paul A. Glossop; Ian A. Nash; Jeremy S. Parker (2008). "Pyridine‐N‐Oxide as a Mild Reoxidant Which Transforms Osmium‐Catalyzed Oxidative Cyclization". Angew. Chem. Int. Ed. doi:10.1002/anie.200705425.
- ↑ Kotohiro Nomura; Shu Zhang (2011). "Design of Vanadium Complex Catalysts for Precise Olefin Polymerization". Chem. Rev. 111: 2342–2362. doi:10.1021/cr100207h.
- ↑ R. A. Miller; R. S. Hoerrner (2003). "Iodine as a Chemoselective Reoxidant of TEMPO: Application to the Oxidation of Alcohols to Aldehydes and Ketones". Org. Lett. 5: 285–287. doi:10.1021/ol0272444.