Related Research Articles

Charcot–Marie–Tooth disease (CMT) is a hereditary motor and sensory neuropathy of the peripheral nervous system characterized by progressive loss of muscle tissue and touch sensation across various parts of the body. This disease is the most commonly inherited neurological disorder, affecting about one in 2,500 people. It is named after those who classically described it: the Frenchman Jean-Martin Charcot (1825–1893), his pupil Pierre Marie (1853–1940), and the Briton Howard Henry Tooth (1856–1925).

Post-polio syndrome is a group of latent symptoms of poliomyelitis (polio), occurring at about a 25–40% rate. These symptoms are caused by the damaging effects of the viral infection on the nervous system. Symptoms typically occur 15 to 30 years after an initial acute paralytic attack. Symptoms include decreasing muscular function or acute weakness with pain and fatigue. The same symptoms may also occur years after a nonparalytic polio (NPP) infection.

A cramp is a sudden, involuntary, painful skeletal muscle contraction or overshortening associated with electrical activity; while generally temporary and non-damaging, they can cause significant pain and a paralysis-like immobility of the affected muscle. A cramp usually goes away on its own over a period of several seconds or (sometimes) minutes. Cramps are common and tend to occur at rest, usually at night. They are also often associated with pregnancy, physical exercise or overexertion, age, in such cases, cramps are called idiopathic, because there is no underlying pathology. In addition to those benign conditions cramps are also associated with many pathological conditions.
Diabetic neuropathy is various types of nerve damage associated with diabetes mellitus. Symptoms depend on the site of nerve damage and can include motor changes such as weakness; sensory symptoms such as numbness, tingling, or pain; or autonomic changes such as urinary symptoms. These changes are thought to result from a microvascular injury involving small blood vessels that supply nerves. Relatively common conditions which may be associated with diabetic neuropathy include distal symmetric polyneuropathy; third, fourth, or sixth cranial nerve palsy; mononeuropathy; mononeuropathy multiplex; diabetic amyotrophy; and autonomic neuropathy.

Mucopolysaccharidoses are a group of metabolic disorders caused by the absence or malfunctioning of lysosomal enzymes needed to break down molecules called glycosaminoglycans (GAGs). These long chains of sugar carbohydrates occur within the cells that help build bone, cartilage, tendons, corneas, skin and connective tissue. GAGs are also found in the fluids that lubricate joints.

Myoclonus is a brief, involuntary, irregular twitching of a muscle, a joint, or a group of muscles, different from clonus, which is rhythmic or regular. Myoclonus describes a medical sign and, generally, is not a diagnosis of a disease. These myoclonic twitches, jerks, or seizures are usually caused by sudden muscle contractions or brief lapses of contraction. The most common circumstance under which they occur is while falling asleep. Myoclonic jerks occur in healthy people and are experienced occasionally by everyone. However, when they appear with more persistence and become more widespread they can be a sign of various neurological disorders. Hiccups are a kind of myoclonic jerk specifically affecting the diaphragm. When a spasm is caused by another person it is known as a provoked spasm. Shuddering attacks in babies fall in this category.
Writer's cramp or focal hand dystonia (FHD) is an idiopathic movement disorder of adult onset, characterized by abnormal posturing and movement of the hand and/or forearm during tasks requiring skilled hand use, such as writing. Overcontraction of affected muscles, cocontraction of agonist and antagonist pairs, and activation of muscles inappropriate to a task all impair use of the affected hand.

Hurler syndrome, also known as mucopolysaccharidosis Type IH (MPS-IH), Hurler's disease, and formerly gargoylism, is a genetic disorder that results in the buildup of large sugar molecules called glycosaminoglycans (GAGs) in lysosomes. The inability to break down these molecules results in a wide variety of symptoms caused by damage to several different organ systems, including but not limited to the nervous system, skeletal system, eyes, and heart.
POEMS syndrome is a rare paraneoplastic syndrome caused by a clone of aberrant plasma cells. The name POEMS is an acronym for some of the disease's major signs and symptoms, as is PEP.

X-linked recessive inheritance is a mode of inheritance in which a mutation in a gene on the X chromosome causes the phenotype to be always expressed in males and in females who are homozygous for the gene mutation, see zygosity. Females with one copy of the mutated gene are carriers.
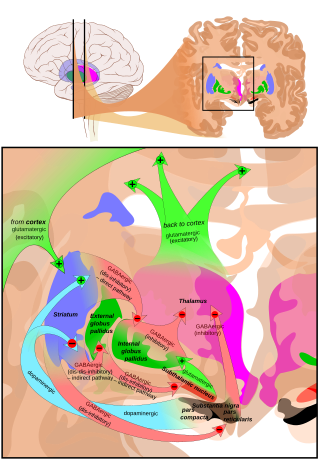
Hyperkinesia refers to an increase in muscular activity that can result in excessive abnormal movements, excessive normal movements, or a combination of both. Hyperkinesia is a state of excessive restlessness which is featured in a large variety of disorders that affect the ability to control motor movement, such as Huntington's disease. It is the opposite of hypokinesia, which refers to decreased bodily movement, as commonly manifested in Parkinson's disease.

Ligamentous laxity, or ligament laxity, is a cause of chronic body pain characterized by loose ligaments. When this condition affects joints in the entire body, it is called generalized joint hypermobility, which occurs in about ten percent of the population, and may be genetic. Loose ligaments can appear in a variety of ways and levels of severity. It also does not always affect the entire body. One could have loose ligaments of the feet, but not of the arms.

Stiff-person syndrome (SPS), also known as stiff-man syndrome, is a rare neurologic disorder of unclear cause characterized by progressive muscular rigidity and stiffness. The stiffness primarily affects the truncal muscles and is superimposed by spasms, resulting in postural deformities. Chronic pain, impaired mobility, and lumbar hyperlordosis are common symptoms.
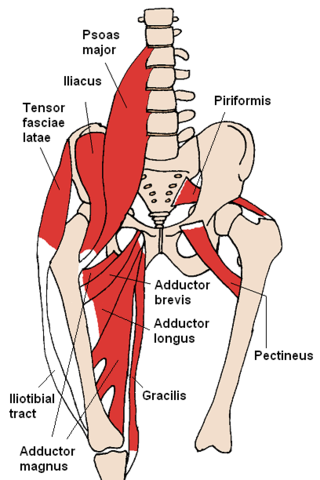
Snapping hip syndrome, also referred to as dancer's hip, is a medical condition characterized by a snapping sensation felt when the hip is flexed and extended. This may be accompanied by a snapping or popping noise and pain or discomfort. Pain often decreases with rest and diminished activity. Snapping hip syndrome is commonly classified by the location of the snapping as either extra-articular or intra-articular.
Chronic inflammatory demyelinating polyneuropathy (CIDP) is an acquired autoimmune disease of the peripheral nervous system characterized by progressive weakness and impaired sensory function in the legs and arms. The disorder is sometimes called chronic relapsing polyneuropathy (CRP) or chronic inflammatory demyelinating polyradiculoneuropathy. CIDP is closely related to Guillain–Barré syndrome and it is considered the chronic counterpart of that acute disease. Its symptoms are also similar to progressive inflammatory neuropathy. It is one of several types of neuropathy.
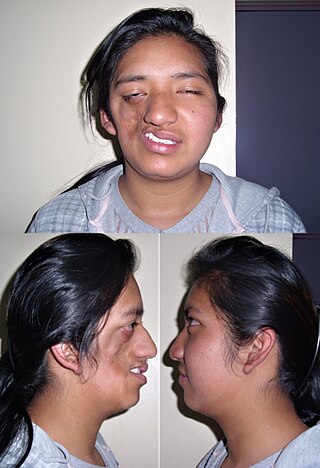
Parry–Romberg syndrome (PRS) is a rare disease characterized by progressive shrinkage and degeneration of the tissues beneath the skin, usually on only one side of the face but occasionally extending to other parts of the body. An autoimmune mechanism is suspected, and the syndrome may be a variant of localized scleroderma, but the precise cause and pathogenesis of this acquired disorder remains unknown. It has been reported in the literature as a possible consequence of sympathectomy. The syndrome has a higher prevalence in females and typically appears between 5 and 15 years of age.
Guillain–Barré syndrome (GBS) is a rapid-onset muscle weakness caused by the immune system damaging the peripheral nervous system. Typically, both sides of the body are involved, and the initial symptoms are changes in sensation or pain often in the back along with muscle weakness, beginning in the feet and hands, often spreading to the arms and upper body. The symptoms may develop over hours to a few weeks. During the acute phase, the disorder can be life-threatening, with about 15% of people developing weakness of the breathing muscles and, therefore, requiring mechanical ventilation. Some are affected by changes in the function of the autonomic nervous system, which can lead to dangerous abnormalities in heart rate and blood pressure.

Camptocormia, also known as bent spine syndrome (BSS), is a symptom of a multitude of diseases that is most commonly seen in the elderly. It is identified by an abnormal thoracolumbar spinal flexion, which is a forward bending of the lower joints of the spine, occurring in a standing position. In order to be classified as BSS, the anterior flexion must be of 45 degrees anteriorly. This classification differentiates it from a similar syndrome known as kyphosis. Although camptocormia is a symptom of many diseases, there are two common origins: neurological and muscular. Camptocormia is treated by alleviating the underlying condition causing it through therapeutic measures or lifestyle changes.
Multifocal motor neuropathy (MMN) is a progressively worsening condition where muscles in the extremities gradually weaken. The disorder, a pure motor neuropathy syndrome, is sometimes mistaken for amyotrophic lateral sclerosis (ALS) because of the similarity in the clinical picture, especially if muscle fasciculations are present. MMN is thought to be autoimmune. It was first described in the mid-1980s.

The HAIR-AN syndrome is a rare subtype of polycystic ovary syndrome (PCOS) characterized by hyperandrogenism (HA), insulin resistance (IR) and acanthosis nigricans (AN). The symptoms of the HAIR-AN syndrome are largely due to severe insulin resistance, which can be secondary to blocking antibodies against the insulin receptor or genetically absent/reduced insulin receptor number/function. Insulin resistance leads to hyperinsulinemia which, in turn, leads to an excess production of androgen hormones by the ovaries. High levels of androgen hormones (hyperandrogenism) in females causes excessive hair growth, acne and irregular menstruation. Patients with both underlying mechanisms of insulin resistance may have more severe hyperandrogenism. Insulin resistance is also associated with diabetes, heart disease and excessive darkening of the skin
References
- ↑ "Archived copy" (PDF). Archived from the original (PDF) on 2005-09-04. Retrieved 2006-08-18.
{{cite web}}: CS1 maint: archived copy as title (link) - ↑ https://www.jstage.jst.go.jp/article/naika1913/91/8/91_8_2325/_pdf(in+Japanese))
- ↑ Satoyoshi syndrome neurologyindia.com
- ↑ Drost G, Verrips A, Hooijkaas H, Zwarts M (March 2004). "Glutamic acid decarboxylase antibodies in Satoyoshi syndrome". Ann. Neurol. 55 (3): 450–1. doi:10.1002/ana.20007. PMID 14991831. S2CID 40178572.