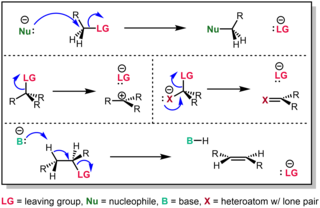
In chemistry, a leaving group is defined by the IUPAC as an atom or group of atoms that detaches from the main or residual part of a substrate during a reaction or elementary step of a reaction. However, in common usage, the term is often limited to a fragment that departs with a pair of electrons in heterolytic bond cleavage. In this usage, a leaving group is a less formal but more commonly used synonym of the term nucleofuge. In this context, leaving groups are generally anions or neutral species, departing from neutral or cationic substrates, respectively, though in rare cases, cations leaving from a dicationic substrate are also known.
In organic chemistry, a carbanion is an anion in which carbon is negatively charged.
Anions that interact weakly with cations are termed non-coordinating anions, although a more accurate term is weakly coordinating anion. Non-coordinating anions are useful in studying the reactivity of electrophilic cations. They are commonly found as counterions for cationic metal complexes with an unsaturated coordination sphere. These special anions are essential components of homogeneous alkene polymerisation catalysts, where the active catalyst is a coordinatively unsaturated, cationic transition metal complex. For example, they are employed as counterions for the 14 valence electron cations [(C5H5)2ZrR]+ (R = methyl or a growing polyethylene chain). Complexes derived from non-coordinating anions have been used to catalyze hydrogenation, hydrosilylation, oligomerization, and the living polymerization of alkenes. The popularization of non-coordinating anions has contributed to increased understanding of agostic complexes wherein hydrocarbons and hydrogen serve as ligands. Non-coordinating anions are important components of many superacids, which result from the combination of Brønsted acids and Lewis acids.

A persistent carbene (also known as stable carbene) is a type of carbene demonstrating particular stability. The best-known examples and by far largest subgroup are the N-heterocyclic carbenes (NHC) (sometimes called Arduengo carbenes), for example diaminocarbenes with the general formula (R2N)2C:, where the four R moieties are typically alkyl and aryl groups. The groups can be linked to give heterocyclic carbenes, such as those derived from imidazole, imidazoline, thiazole or triazole.

Fluoroantimonic acid is a mixture of hydrogen fluoride and antimony pentafluoride, containing various cations and anions. This mixture is a superacid that, in some sense, is over a billion times stronger than 100% sulfuric acid in terms of its protonating ability measured by Hammett function. It even protonates some hydrocarbons to afford pentacoordinate carbocations. Fluoroantimonic acid is corrosive. Like its precursor hydrogen fluoride, it attacks glass, but can be stored in containers lined with PTFE (Teflon) or PFA.

Tetrafluoroborate is the anion BF−
4. This tetrahedral species is isoelectronic with tetrafluoroberyllate (BeF2−
4), tetrafluoromethane (CF4), and tetrafluoroammonium (NF+
4) and is valence isoelectronic with many stable and important species including the perchlorate anion, ClO−
4, which is used in similar ways in the laboratory. It arises by the reaction of fluoride salts with the Lewis acid BF3, treatment of tetrafluoroboric acid with base, or by treatment of boric acid with hydrofluoric acid.

Hexafluorophosphate is an anion with chemical formula of [PF6]−. It is an octahedral species that imparts no color to its salts. [PF6]− is isoelectronic with sulfur hexafluoride, SF6, and the hexafluorosilicate dianion, [SiF6]2−, and hexafluoroantimonate [SbF6]−. In this anion, phosphorus has a valence of 5. Being poorly nucleophilic, hexafluorophosphate is classified as a non-coordinating anion.
In chemistry, ion association is a chemical reaction whereby ions of opposite electric charge come together in solution to form a distinct chemical entity. Ion associates are classified, according to the number of ions that associate with each other, as ion pairs, ion triplets, etc. Ion pairs are also classified according to the nature of the interaction as contact, solvent-shared or solvent-separated. The most important factor to determine the extent of ion association is the dielectric constant of the solvent. Ion associates have been characterized by means of vibrational spectroscopy, as introduced by Niels Bjerrum, and dielectric-loss spectroscopy.

Tetrakis[3,5-bis(trifluoromethyl)phenyl]borate is an anion with chemical formula [{3,5-(CF3)2C6H3}4B]−, which is commonly abbreviated as [BArF4]−, indicating the presence of fluorinated aryl (ArF) groups. It is sometimes referred to as Kobayashi's anion in honour of Hiroshi Kobayashi who led the team that first synthesised it. More commonly it is affectionately nicknamed "BARF." The BARF ion is also abbreviated BArF24−, to distinguish it from the closely related BArF−
20, [(C6F5)4B]−.
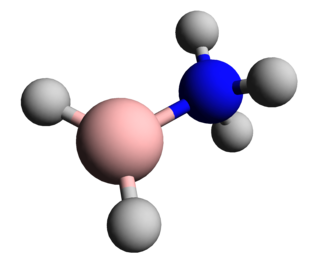
In chemistry, a boranylium ion is an inorganic cation with the chemical formula BR+
2, where R represents a non-specific substituent. Being electron-deficient, boranylium ions form adducts with Lewis bases. Boranylium ions have historical names that depend on the number of coordinated ligands:

Brookhart's acid is the salt of the diethyl ether oxonium ion and tetrakis[3,5-bis(trifluoromethyl)phenyl]borate (BAr′4). It is a colorless solid, used as a strong acid. The compound was first reported by Volpe, Grant, and Brookhart in 1992.
In chemistry, the Gutmann–Beckett method is an experimental procedure used by chemists to assess the Lewis acidity of molecular species. Triethylphosphine oxide is used as a probe molecule and systems are evaluated by 31P-NMR spectroscopy. In 1975, Viktor Gutmann used 31P-NMR spectroscopy to parameterize Lewis acidity of solvents by acceptor numbers (AN). In 1996, Michael A. Beckett recognised its more generally utility and adapted the procedure so that it could be easily applied to molecular species, when dissolved in weakly Lewis acidic solvents. The term Gutmann–Beckett method was first used in chemical literature in 2007.
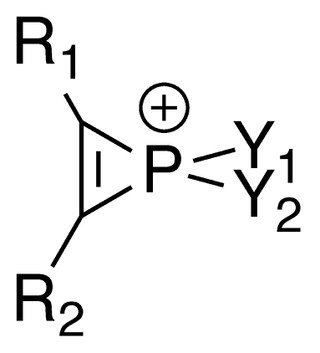
Phosphirenium ions are a series of organophosphorus compounds containing unsaturated three-membered ring phosphorus (V) heterocycles and σ*-aromaticity is believed to be present in such molecules. Many of the salts containing phosphirenium ions have been isolated and characterized by NMR spectroscopy and X-ray crystallography.

Silylones are a class of zero-valent monatomic silicon complexes, characterized as having two lone pairs and two donor-acceptor ligand interactions stabilizing a silicon(0) center. Synthesis of silylones generally involves the use of sterically bulky carbenes to stabilize highly reactive Si(0) centers. For this reason, silylones are sometimes referred to siladicarbenes. To date, silylones have been synthesized with cyclic alkyl amino carbenes (cAAC) and bidentate N-heterocyclic carbenes (bis-NHC). They are capable of reactions with a variety of substrates, including chalcogens and carbon dioxide.

An N-Heterocyclic silylene (NHSi) is an uncharged heterocyclic chemical compound consisting of a divalent silicon atom bonded to two nitrogen atoms. The isolation of the first stable NHSi, also the first stable dicoordinate silicon compound, was reported in 1994 by Michael Denk and Robert West three years after Anthony Arduengo first isolated an N-heterocyclic carbene, the lighter congener of NHSis. Since their first isolation, NHSis have been synthesized and studied with both saturated and unsaturated central rings ranging in size from 4 to 6 atoms. The stability of NHSis, especially 6π aromatic unsaturated five-membered examples, make them useful systems to study the structure and reactivity of silylenes and low-valent main group elements in general. Though not used outside of academic settings, complexes containing NHSis are known to be competent catalysts for industrially important reactions. This article focuses on the properties and reactivity of five-membered NHSis.

Ge(II) dicationic complexes refer to coordination compounds of germanium with a +2 formal oxidation state, and a +2 charge on the overall complex. In some of these coordination complexes, the coordination is strongly ionic, localizing a +2 charge on Ge, while in others the bonding is more covalent, delocalizing the cationic charge away from Ge. Examples of dicationic Ge(II) complexes are much rarer than monocationic Ge(II) complexes, often requiring the use of bulky ligands to shield the germanium center. Dicationic complexes of Ge(II) have been isolated with bulky isocyanide and carbene ligands. Much more weakly coordinated Germanium (II) dications have been isolated as complexes with polyether ligands, such as crown ethers and [2.2.2]cryptand. Crown ethers and cryptands are typically known for their ability to bind metal cations, however these ligands have also been employed in stabilizing low-valent cations of heavier p-block elements. A Ge2+ ion's valence shell consists of a filled valence s orbital but empty valence p orbitals, giving rise to atypical bonding in these complexes. Germanium is a metalloid of the carbon group, typically forming compounds with mainly covalent bonding, contrasting with the dative bonding observed in these coordination complexes.
Gallium monoiodide (GaI or Ga4I4) is a low-valent gallium species that acts as a reactive intermediate for many gallium-based products. Gallium(I) halides were first crystallographically characterized by Schnöckel and coworkers and have allowed a synthetic route to many low-valent gallium species. However, chemical syntheses that employ “GaI” rather than gallium(I) halide precursors have been increasingly investigated given the ease of synthesis of this reagent. While the synthetic method of Schnöckel and coworkers to synthesize gallium(I) halides require extraordinarily high temperatures, the straightforward preparation of “GaI” at near room temperature has allowed for the exploration of new gallium-based chemistries.

Polyfluoroalkoxyaluminates (PFAA) are weakly coordinating anions many of which are of the form [Al(ORF)4]−. Most PFAA's possesses an Al(III) center coordinated by four −ORF (RF = -CPh(CF3)2 (hfpp), -CH(CF3)2 (hfip), -C(CH3)(CF3)2 (hftb), -C(CF3)3 (pftb)) ligands, giving the anion an overall -1 charge. The most weakly coordinating PFAA is an aluminate dimer, [F{Al(Opftb)3}2]−, which possess a bridging fluoride between two Al(III) centers. The first PFAA, [Al(Ohfpp)4]−, was synthesized in 1996 by Steven Strauss, and several other analogs have since been synthesized, including [Al(Ohfip)4]−, [Al(Ohftb)4]−, and [Al(Opftb)4]− by Ingo Krossing in 2001. These chemically inert and very weakly coordinating ions have been used to stabilize unusual cations, isolate reactive species, and synthesize strong Brønsted acids.

Superelectrophilic anions are a class of molecular ions that exhibit highly electrophilic reaction behavior despite their overall negative charge. Thus, they are even able to bind the unreactive noble gases or molecular nitrogen at room temperature. The only representatives known so far are the fragment ions of the type [B12X11]– derived from the closo-dodecaborate dianions [B12X12]2–. X represents a substituent connected to a boron atom (cf. Fig. 1). For this reason, the following article deals exclusively with superelectrophilic anions of this type.
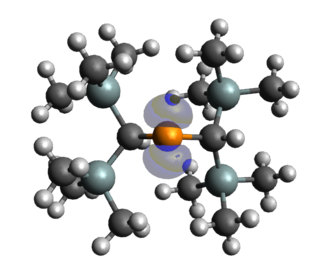
Stable and persistent phosphorus radicals are phosphorus-centred radicals that are isolable and can exist for at least short periods of time. Radicals consisting of main group elements are often very reactive and undergo uncontrollable reactions, notably dimerization and polymerization. The common strategies for stabilising these phosphorus radicals usually include the delocalisation of the unpaired electron over a pi system or nearby electronegative atoms, and kinetic stabilisation with bulky ligands. Stable and persistent phosphorus radicals can be classified into three categories: neutral, cationic, and anionic radicals. Each of these classes involve various sub-classes, with neutral phosphorus radicals being the most extensively studied. Phosphorus exists as one isotope 31P (I = 1/2) with large hyperfine couplings relative to other spin active nuclei, making phosphorus radicals particularly attractive for spin-labelling experiments.
