
Elias James Corey is an American organic chemist. In 1990, he won the Nobel Prize in Chemistry "for his development of the theory and methodology of organic synthesis", specifically retrosynthetic analysis. Regarded by many as one of the greatest living chemists, he has developed numerous synthetic reagents, methodologies and total syntheses and has advanced the science of organic synthesis considerably.
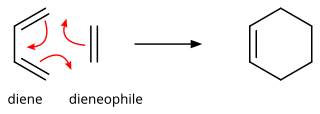
In organic chemistry, the Diels–Alder reaction is a chemical reaction between a conjugated diene and a substituted alkene, commonly termed the dienophile, to form a substituted cyclohexene derivative. It is the prototypical example of a pericyclic reaction with a concerted mechanism. More specifically, it is classified as a thermally-allowed [4+2] cycloaddition with Woodward–Hoffmann symbol [π4s + π2s]. It was first described by Otto Diels and Kurt Alder in 1928. For the discovery of this reaction, they were awarded the Nobel Prize in Chemistry in 1950. Through the simultaneous construction of two new carbon–carbon bonds, the Diels–Alder reaction provides a reliable way to form six-membered rings with good control over the regio- and stereochemical outcomes. Consequently, it has served as a powerful and widely applied tool for the introduction of chemical complexity in the synthesis of natural products and new materials. The underlying concept has also been applied to π-systems involving heteroatoms, such as carbonyls and imines, which furnish the corresponding heterocycles; this variant is known as the hetero-Diels–Alder reaction. The reaction has also been generalized to other ring sizes, although none of these generalizations have matched the formation of six-membered rings in terms of scope or versatility. Because of the negative values of ΔH° and ΔS° for a typical Diels–Alder reaction, the microscopic reverse of a Diels–Alder reaction becomes favorable at high temperatures, although this is of synthetic importance for only a limited range of Diels-Alder adducts, generally with some special structural features; this reverse reaction is known as the retro-Diels–Alder reaction.
David A. Evans was an American chemist who was the Abbott and James Lawrence professor of chemistry at Harvard University. He was a prominent figure in the field of organic chemistry and his research focused on synthetic chemistry and total synthesis, particularly of large biologically active molecules. Among his best-known work is the development of aldol reaction methodology.

In biochemistry and pharmacology, a ligand is a substance that forms a complex with a biomolecule to serve a biological purpose. The etymology stems from ligare, which means 'to bind'. In protein-ligand binding, the ligand is usually a molecule which produces a signal by binding to a site on a target protein. The binding typically results in a change of conformational isomerism (conformation) of the target protein. In DNA-ligand binding studies, the ligand can be a small molecule, ion, or protein which binds to the DNA double helix. The relationship between ligand and binding partner is a function of charge, hydrophobicity, and molecular structure.
Ring-closing metathesis (RCM) is a widely used variation of olefin metathesis in organic chemistry for the synthesis of various unsaturated rings via the intramolecular metathesis of two terminal alkenes, which forms the cycloalkene as the E- or Z- isomers and volatile ethylene.

2-Imidazoline (Preferred IUPAC name: 4,5-dihydro-1H-imidazole) is one of three isomers of the nitrogen-containing heterocycle imidazoline, with the formula C3H6N2. The 2-imidazolines are the most common imidazolines commercially, as the ring exists in some natural products and some pharmaceuticals. They also have been examined in the context of organic synthesis, coordination chemistry, and homogeneous catalysis.
The Nazarov cyclization reaction is a chemical reaction used in organic chemistry for the synthesis of cyclopentenones. The reaction is typically divided into classical and modern variants, depending on the reagents and substrates employed. It was originally discovered by Ivan Nikolaevich Nazarov (1906–1957) in 1941 while studying the rearrangements of allyl vinyl ketones.
Martin Gerhardt Banwell, Hon.FRSNZ is an organic chemist specialising in biotransformations and natural product synthesis.
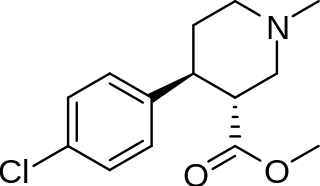
(+)-CPCA is a stimulant drug similar in structure to pethidine and to RTI-31, but nocaine is lacking the two-carbon bridge of RTI-31's tropane skeleton. This compound was first developed as a substitute agent for cocaine.

Larry E. Overman is Distinguished Professor of Chemistry at the University of California, Irvine. He was born in Chicago in 1943. Overman obtained a B.A. degree from Earlham College in 1965, and he completed his Ph.D. in chemistry from the University of Wisconsin–Madison in 1969, under Howard Whitlock Jr. Professor Overman is a member of the United States National Academy of Sciences and the American Academy of Arts and Sciences. He was the recipient of the Arthur C. Cope Award in 2003, and he was awarded the Tetrahedron Prize for Creativity in Organic Chemistry for 2008.
Diazirines are a class of organic molecules consisting of a carbon bound to two nitrogen atoms, which are double-bonded to each other, forming a cyclopropene-like ring, 3H-diazirene. They are isomeric with diazocarbon groups, and like them can serve as precursors for carbenes by loss of a molecule of dinitrogen. For example, irradiation of diazirines with ultraviolet light leads to carbene insertion into various C-H, N-H, and O-H bonds. Hence, diazirines have grown in popularity as small photo-reactive crosslinking reagents. They are often used in photoaffinity labeling studies to observe a variety of interactions, including ligand-receptor, ligand-enzyme, protein-protein, and protein-nucleic acid interactions.

Dynamic combinatorial chemistry (DCC); also known as constitutional dynamic chemistry (CDC) is a method to the generation of new molecules formed by reversible reaction of simple building blocks under thermodynamic control. The library of these reversibly interconverting building blocks is called a dynamic combinatorial library (DCL). All constituents in a DCL are in equilibrium, and their distribution is determined by their thermodynamic stability within the DCL. The interconversion of these building blocks may involve covalent or non-covalent interactions. When a DCL is exposed to an external influence, the equilibrium shifts and those components that interact with the external influence are stabilised and amplified, allowing more of the active compound to be formed.

Yoshito Kishi is a Japanese chemist who is the Morris Loeb Professor of Chemistry at Harvard University. He is known for his contributions to the sciences of organic synthesis and total synthesis.
The Buchner ring expansion is a two-step organic C-C bond forming reaction used to access 7-membered rings. The first step involves formation of a carbene from ethyl diazoacetate, which cyclopropanates an aromatic ring. The ring expansion occurs in the second step, with an electrocyclic reaction opening the cyclopropane ring to form the 7-membered ring.
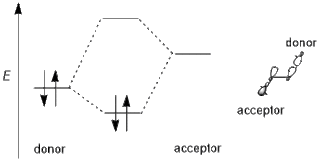
In chemistry, primarily organic and computational chemistry, a stereoelectronic effect is an effect on molecular geometry, reactivity, or physical properties due to spatial relationships in the molecules' electronic structure, in particular the interaction between atomic and/or molecular orbitals. Phrased differently, stereoelectronic effects can also be defined as the geometric constraints placed on the ground and/or transition states of molecules that arise from considerations of orbital overlap. Thus, a stereoelectronic effect explains a particular molecular property or reactivity by invoking stabilizing or destabilizing interactions that depend on the relative orientations of electrons in space.
Rearrangements, especially those that can participate in cascade reactions, such as the aza-Cope rearrangements, are of high practical as well as conceptual importance in organic chemistry, due to their ability to quickly build structural complexity out of simple starting materials. The aza-Cope rearrangements are examples of heteroatom versions of the Cope rearrangement, which is a [3,3]-sigmatropic rearrangement that shifts single and double bonds between two allylic components. In accordance with the Woodward-Hoffman rules, thermal aza-Cope rearrangements proceed suprafacially. Aza-Cope rearrangements are generally classified by the position of the nitrogen in the molecule :
In organic chemistry, the oxy-Cope rearrangement is a chemical reaction. It involves reorganization of the skeleton of certain unsaturated alcohols. It is a variation of the Cope rearrangement in which 1,5-dien-3-ols are converted to unsaturated carbonyl compounds by a mechanism typical for such a [3,3]-sigmatropic rearrangement.

The Mizoroki−Heck coupling of aryl halides and alkenes to form C(sp2)–C(sp2) bonds has become a staple transformation in organic synthesis, owing to its broad functional group compatibility and varied scope. In stark contrast, the palladium-catalyzed reductive Heck reaction has received considerably less attention, despite the fact that early reports of this reaction date back almost half a century. From the perspective of retrosynthetic logic, this transformation is highly enabling because it can forge alkyl–aryl linkages from widely available alkenes, rather than from the less accessible and/or more expensive alkyl halide or organometallic C(sp3) synthons that are needed in a classical aryl/alkyl cross-coupling.
Balaram Mukhopadhyay is an Indian Bengali carbohydrate chemist and a professor at the department of chemical sciences of the Indian Institute of Science Education and Research, Kolkata. Balaram is mainly known for his work in the field of synthetic carbohydrate chemistry. He was given the Excellence in Carbohydrate Research Award by the Association of Carbohydrate Chemists and Technologists India (ACCTI) in 2018 for his contribution towards field of carbohydrates.

Teruaki Mukaiyama was a Japanese organic chemist. One of the most prolific chemists of the 20th century in the field of organic synthesis, Mukaiyama helped establish the field of organic chemistry in Japan after World War II.
