In biology, a mutation is an alteration in the nucleic acid sequence of the genome of an organism, virus, or extrachromosomal DNA. Viral genomes contain either DNA or RNA. Mutations result from errors during DNA or viral replication, mitosis, or meiosis or other types of damage to DNA, which then may undergo error-prone repair, cause an error during other forms of repair, or cause an error during replication. Mutations may also result from insertion or deletion of segments of DNA due to mobile genetic elements.

The human genome is a complete set of nucleic acid sequences for humans, encoded as DNA within the 23 chromosome pairs in cell nuclei and in a small DNA molecule found within individual mitochondria. These are usually treated separately as the nuclear genome and the mitochondrial genome. Human genomes include both protein-coding DNA sequences and various types of DNA that does not encode proteins. The latter is a diverse category that includes DNA coding for non-translated RNA, such as that for ribosomal RNA, transfer RNA, ribozymes, small nuclear RNAs, and several types of regulatory RNAs. It also includes promoters and their associated gene-regulatory elements, DNA playing structural and replicatory roles, such as scaffolding regions, telomeres, centromeres, and origins of replication, plus large numbers of transposable elements, inserted viral DNA, non-functional pseudogenes and simple, highly repetitive sequences. Introns make up a large percentage of non-coding DNA. Some of this non-coding DNA is non-functional junk DNA, such as pseudogenes, but there is no firm consensus on the total amount of junk DNA.
An Alu element is a short stretch of DNA originally characterized by the action of the Arthrobacter luteus (Alu) restriction endonuclease. Alu elements are the most abundant transposable elements, containing over one million copies dispersed throughout the human genome. Alu elements were thought to be selfish or parasitic DNA, because their sole known function is self reproduction. However, they are likely to play a role in evolution and have been used as genetic markers. They are derived from the small cytoplasmic 7SL RNA, a component of the signal recognition particle. Alu elements are highly conserved within primate genomes and originated in the genome of an ancestor of Supraprimates.
Gene duplication is a major mechanism through which new genetic material is generated during molecular evolution. It can be defined as any duplication of a region of DNA that contains a gene. Gene duplications can arise as products of several types of errors in DNA replication and repair machinery as well as through fortuitous capture by selfish genetic elements. Common sources of gene duplications include ectopic recombination, retrotransposition event, aneuploidy, polyploidy, and replication slippage.

Haploinsufficiency in genetics describes a model of dominant gene action in diploid organisms, in which a single copy of the wild-type allele at a locus in heterozygous combination with a variant allele is insufficient to produce the wild-type phenotype. Haploinsufficiency may arise from a de novo or inherited loss-of-function mutation in the variant allele, such that it yields little or no gene product. Although the other, standard allele still produces the standard amount of product, the total product is insufficient to produce the standard phenotype. This heterozygous genotype may result in a non- or sub-standard, deleterious, and (or) disease phenotype. Haploinsufficiency is the standard explanation for dominant deleterious alleles.
Indel (insertion-deletion) is a molecular biology term for an insertion or deletion of bases in the genome of an organism. Indels ≥ 50 bases in length are classified as structural variants.

Copy number variation (CNV) is a phenomenon in which sections of the genome are repeated and the number of repeats in the genome varies between individuals. Copy number variation is a type of structural variation: specifically, it is a type of duplication or deletion event that affects a considerable number of base pairs. Approximately two-thirds of the entire human genome may be composed of repeats and 4.8–9.5% of the human genome can be classified as copy number variations. In mammals, copy number variations play an important role in generating necessary variation in the population as well as disease phenotype.

Human genetic variation is the genetic differences in and among populations. There may be multiple variants of any given gene in the human population (alleles), a situation called polymorphism.

The 1000 Genomes Project, launched in January 2008, was an international research effort to establish by far the most detailed catalogue of human genetic variation. Scientists planned to sequence the genomes of at least one thousand anonymous participants from a number of different ethnic groups within the following three years, using newly developed technologies which were faster and less expensive. In 2010, the project finished its pilot phase, which was described in detail in a publication in the journal Nature. In 2012, the sequencing of 1092 genomes was announced in a Nature publication. In 2015, two papers in Nature reported results and the completion of the project and opportunities for future research.
Low copy repeats (LCRs), also known as segmental duplications (SDs), are DNA sequences present in multiple locations within a genome that share high levels of sequence identity.
Koolen–De Vries syndrome (KdVS), also known as 17q21.31 microdeletion syndrome, is a rare genetic disorder caused by a deletion of a segment of chromosome 17 which contains six genes. This deletion syndrome was discovered independently in 2006 by three different research groups.
DECIPHER is a web-based resource and database of genomic variation data from analysis of patient DNA. It documents submicroscopic chromosome abnormalities and pathogenic sequence variants, from over 25000 patients and maps them to the human genome using Ensembl or UCSC Genome Browser. In addition it catalogues the clinical characteristics from each patient and maintains a database of microdeletion/duplication syndromes, together with links to relevant scientific reports and support groups.
8p23.1 duplication syndrome is a rare genetic disorder caused by a duplication of a region from human chromosome 8. This duplication syndrome has an estimated prevalence of 1 in 64,000 births and is the reciprocal of the 8p23.1 deletion syndrome. The 8p23.1 duplication is associated with a variable phenotype including one or more of speech delay, developmental delay, mild dysmorphism, with prominent forehead and arched eyebrows, and congenital heart disease (CHD).
Non-allelic homologous recombination (NAHR) is a form of homologous recombination that occurs between two lengths of DNA that have high sequence similarity, but are not alleles.
esophageal candidiasis1q21.1 deletion syndrome is a rare aberration of chromosome 1. A human cell has one pair of identical chromosomes on chromosome 1. With the 1q21.1 deletion syndrome, one chromosome of the pair is not complete, because a part of the sequence of the chromosome is missing. One chromosome has the normal length and the other is too short.

Charles Lee is Director and Professor of The Jackson Laboratory for Genomic Medicine, The Robert Alvine Family Endowed Chair and a board certified clinical cytogeneticist who has an active research program in the identification and characterization of structural genomic variants using advanced technology platforms. His laboratory was the first to describe genome-wide structural genomic variants among humans with the subsequent development of two human CNV maps that are now actively used in the diagnoses of array based genetic tests. Lee is also currently the President of the Human Genome Organisation (HUGO).

End-sequence profiling (ESP) is a method based on sequence-tagged connectors developed to facilitate de novo genome sequencing to identify high-resolution copy number and structural aberrations such as inversions and translocations.
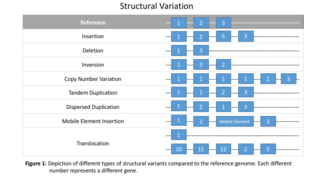
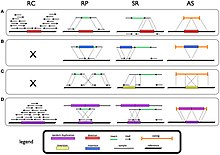
Structural variation in the human genome is operationally defined as genomic alterations, varying between individuals, that involve DNA segments larger than 1 kilo base (kb), and could be either microscopic or submicroscopic. This definition distinguishes them from smaller variants that are less than 1 kb in size such as short deletions, insertions, and single nucleotide variants.
ANNOVAR is a bioinformatics software tool for the interpretation and prioritization of single nucleotide variants (SNVs), insertions, deletions, and copy number variants (CNVs) of a given genome.
Human somatic variations are somatic mutations both at early stages of development and in adult cells. These variations can lead either to pathogenic phenotypes or not, even if their function in healthy conditions is not completely clear yet.