
A DNA microarray is a collection of microscopic DNA spots attached to a solid surface. Scientists use DNA microarrays to measure the expression levels of large numbers of genes simultaneously or to genotype multiple regions of a genome. Each DNA spot contains picomoles of a specific DNA sequence, known as probes. These can be a short section of a gene or other DNA element that are used to hybridize a cDNA or cRNA sample under high-stringency conditions. Probe-target hybridization is usually detected and quantified by detection of fluorophore-, silver-, or chemiluminescence-labeled targets to determine relative abundance of nucleic acid sequences in the target. The original nucleic acid arrays were macro arrays approximately 9 cm × 12 cm and the first computerized image based analysis was published in 1981. It was invented by Patrick O. Brown. An example of its application is in SNPs arrays for polymorphisms in cardiovascular diseases, cancer, pathogens and GWAS analysis. Also for identification of structural variations and measurement of gene expression.

Regulation of gene expression, or gene regulation, includes a wide range of mechanisms that are used by cells to increase or decrease the production of specific gene products. Sophisticated programs of gene expression are widely observed in biology, for example to trigger developmental pathways, respond to environmental stimuli, or adapt to new food sources. Virtually any step of gene expression can be modulated, from transcriptional initiation, to RNA processing, and to the post-translational modification of a protein. Often, one gene regulator controls another, and so on, in a gene regulatory network.

DNA-binding proteins are proteins that have DNA-binding domains and thus have a specific or general affinity for single- or double-stranded DNA. Sequence-specific DNA-binding proteins generally interact with the major groove of B-DNA, because it exposes more functional groups that identify a base pair. However, there are some known minor groove DNA-binding ligands such as netropsin, distamycin, Hoechst 33258, pentamidine, DAPI and others.
Comparative genomic hybridization(CGH) is a molecular cytogenetic method for analysing copy number variations (CNVs) relative to ploidy level in the DNA of a test sample compared to a reference sample, without the need for culturing cells. The aim of this technique is to quickly and efficiently compare two genomic DNA samples arising from two sources, which are most often closely related, because it is suspected that they contain differences in terms of either gains or losses of either whole chromosomes or subchromosomal regions. This technique was originally developed for the evaluation of the differences between the chromosomal complements of solid tumor and normal tissue, and has an improved resolution of 5–10 megabases compared to the more traditional cytogenetic analysis techniques of giemsa banding and fluorescence in situ hybridization (FISH) which are limited by the resolution of the microscope utilized.

Functional genomics is a field of molecular biology that attempts to describe gene functions and interactions. Functional genomics make use of the vast data generated by genomic and transcriptomic projects. Functional genomics focuses on the dynamic aspects such as gene transcription, translation, regulation of gene expression and protein–protein interactions, as opposed to the static aspects of the genomic information such as DNA sequence or structures. A key characteristic of functional genomics studies is their genome-wide approach to these questions, generally involving high-throughput methods rather than a more traditional “gene-by-gene” approach.
The transcriptome is the set of all RNA transcripts, including coding and non-coding, in an individual or a population of cells. The term can also sometimes be used to refer to all RNAs, or just mRNA, depending on the particular experiment. The term transcriptome is a portmanteau of the words transcript and genome; it is associated with the process of transcript production during the biological process of transcription.

Fluorescence in situ hybridization (FISH) is a molecular cytogenetic technique that uses fluorescent probes that bind to only those parts of a nucleic acid sequence with a high degree of sequence complementarity. It was developed by biomedical researchers in the early 1980s to detect and localize the presence or absence of specific DNA sequences on chromosomes. Fluorescence microscopy can be used to find out where the fluorescent probe is bound to the chromosomes. FISH is often used for finding specific features in DNA for use in genetic counseling, medicine, and species identification. FISH can also be used to detect and localize specific RNA targets in cells, circulating tumor cells, and tissue samples. In this context, it can help define the spatial-temporal patterns of gene expression within cells and tissues.
A DNA-binding domain (DBD) is an independently folded protein domain that contains at least one structural motif that recognizes double- or single-stranded DNA. A DBD can recognize a specific DNA sequence or have a general affinity to DNA. Some DNA-binding domains may also include nucleic acids in their folded structure.

A nuclear run-on assay is conducted to identify the genes that are being transcribed at a certain time point. Approximately one million cell nuclei are isolated and incubated with labeled nucleotides, and genes in the process of being transcribed are detected by hybridization of extracted RNA to gene specific probes on a blot. Garcia-Martinez et al. (2004) developed a protocol for the yeast S. cerevisiae that allows for the calculation of transcription rates (TRs) for all yeast genes to estimate mRNA stabilities for all yeast mRNAs.
An RNA spike-in is an RNA transcript of known sequence and quantity used to calibrate measurements in RNA hybridization assays, such as DNA microarray experiments, RT-qPCR, and RNA-Seq.

Molecular cytogenetics combines two disciplines, molecular biology and cytogenetics, and involves the analyzation of chromosome structure to help distinguish normal and cancer-causing cells. Human cytogenetics began in 1956 when it was discovered that normal human cells contain 46 chromosomes. However, the first microscopic observations of chromosomes were reported by Arnold, Flemming, and Hansemann in the late 1800s. Their work was ignored for decades until the actual chromosome number in humans was discovered as 46. In 1879, Arnold examined sarcoma and carcinoma cells having very large nuclei. Today, the study of molecular cytogenetics can be useful in diagnosing and treating various malignancies such as hematological malignancies, brain tumors, and other precursors of cancer. The field is overall focused on studying the evolution of chromosomes, more specifically the number, structure, function, and origin of chromosome abnormalities. It includes a series of techniques referred to as fluorescence in situ hybridization, or FISH, in which DNA probes are labeled with different colored fluorescent tags to visualize one or more specific regions of the genome. Introduced in the 1980s, FISH uses probes with complementary base sequences to locate the presence or absence of the specific DNA regions you are looking for. FISH can either be performed as a direct approach to metaphase chromosomes or interphase nuclei. Alternatively, an indirect approach can be taken in which the entire genome can be assessed for copy number changes using virtual karyotyping. Virtual karyotypes are generated from arrays made of thousands to millions of probes, and computational tools are used to recreate the genome in silico.
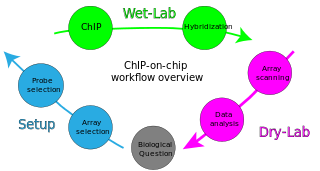
ChIP-on-chip is a technology that combines chromatin immunoprecipitation ('ChIP') with DNA microarray ("chip"). Like regular ChIP, ChIP-on-chip is used to investigate interactions between proteins and DNA in vivo. Specifically, it allows the identification of the cistrome, the sum of binding sites, for DNA-binding proteins on a genome-wide basis. Whole-genome analysis can be performed to determine the locations of binding sites for almost any protein of interest. As the name of the technique suggests, such proteins are generally those operating in the context of chromatin. The most prominent representatives of this class are transcription factors, replication-related proteins, like origin recognition complex protein (ORC), histones, their variants, and histone modifications.
ChIP-sequencing, also known as ChIP-seq, is a method used to analyze protein interactions with DNA. ChIP-seq combines chromatin immunoprecipitation (ChIP) with massively parallel DNA sequencing to identify the binding sites of DNA-associated proteins. It can be used to map global binding sites precisely for any protein of interest. Previously, ChIP-on-chip was the most common technique utilized to study these protein–DNA relations.
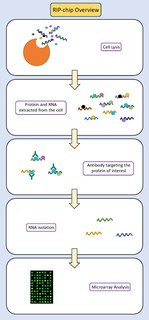
RIP-chip is a molecular biology technique which combines RNA immunoprecipitation with a microarray. The purpose of this technique is to identify which RNA sequences interact with a particular RNA binding protein of interest in vivo. It can also be used to determine relative levels of gene expression, to identify subsets of RNAs which may be co-regulated, or to identify RNAs that may have related functions. This technique provides insight into the post-transcriptional gene regulation which occurs between RNA and RNA binding proteins.
Epigenomics is the study of the complete set of epigenetic modifications on the genetic material of a cell, known as the epigenome. The field is analogous to genomics and proteomics, which are the study of the genome and proteome of a cell. Epigenetic modifications are reversible modifications on a cell's DNA or histones that affect gene expression without altering the DNA sequence. Epigenomic maintenance is a continuous process and plays an important role in stability of eukaryotic genomes by taking part in crucial biological mechanisms like DNA repair. Plant flavones are said to be inhibiting epigenomic marks that cause cancers. Two of the most characterized epigenetic modifications are DNA methylation and histone modification. Epigenetic modifications play an important role in gene expression and regulation, and are involved in numerous cellular processes such as in differentiation/development and tumorigenesis. The study of epigenetics on a global level has been made possible only recently through the adaptation of genomic high-throughput assays.
Methylated DNA immunoprecipitation is a large-scale purification technique in molecular biology that is used to enrich for methylated DNA sequences. It consists of isolating methylated DNA fragments via an antibody raised against 5-methylcytosine (5mC). This technique was first described by Weber M. et al. in 2005 and has helped pave the way for viable methylome-level assessment efforts, as the purified fraction of methylated DNA can be input to high-throughput DNA detection methods such as high-resolution DNA microarrays (MeDIP-chip) or next-generation sequencing (MeDIP-seq). Nonetheless, understanding of the methylome remains rudimentary; its study is complicated by the fact that, like other epigenetic properties, patterns vary from cell-type to cell-type.
Copy number analysis usually refers to the process of analyzing data produced by a test for DNA copy number variation in patient's sample. Such analysis helps detect chromosomal copy number variation that may cause or may increase risks of various critical disorders. Copy number variation can be detected with various types of tests such as fluorescent in situ hybridization, comparative genomic hybridization and with high-resolution array-based tests based on array comparative genomic hybridization, SNP array technologies and high resolution microarrays that include copy number probes as well an SNPs. Array-based methods have been accepted as the most efficient in terms of their resolution and high-throughput nature and the highest coverage and they are also referred to as virtual karyotype. Data analysis for an array-based DNA copy number test can be very challenging though due to very high volume of data that come out of an array platform.
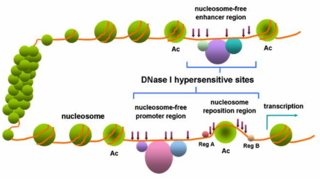
In genetics, DNase I hypersensitive sites (DHSs) are regions of chromatin that are sensitive to cleavage by the DNase I enzyme. In these specific regions of the genome, chromatin has lost its condensed structure, exposing the DNA and making it accessible. This raises the availability of DNA to degradation by enzymes, such as DNase I. These accessible chromatin zones are functionally related to transcriptional activity, since this remodeled state is necessary for the binding of proteins such as transcription factors.
Transcriptomics technologies are the techniques used to study an organism's transcriptome, the sum of all of its RNA transcripts. The information content of an organism is recorded in the DNA of its genome and expressed through transcription. Here, mRNA serves as a transient intermediary molecule in the information network, whilst non-coding RNAs perform additional diverse functions. A transcriptome captures a snapshot in time of the total transcripts present in a cell. Transcriptomics technologies provide a broad account of which cellular processes are active and which are dormant. A major challenge in molecular biology lies in understanding how the same genome can give rise to different cell types and how gene expression is regulated.

MNase-seq, short for micrococcal nuclease digestion with deep sequencing, is a molecular biological technique that was first pioneered in 2006 to measure nucleosome occupancy in the C. elegans genome, and was subsequently applied to the human genome in 2008. Though, the term ‘MNase-seq’ had not been coined until a year later, in 2009. Briefly, this technique relies on the use of the non-specific endo-exonuclease micrococcal nuclease, an enzyme derived from the bacteria Staphylococcus aureus, to bind and cleave protein-unbound regions of DNA on chromatin. DNA bound to histones or other chromatin-bound proteins may remain undigested. The uncut DNA is then purified from the proteins and sequenced through one or more of the various Next-Generation sequencing methods.





