Related Research Articles

Macrocephaly is a condition in which circumference of the human head is abnormally large. It may be pathological or harmless, and can be a familial genetic characteristic. People diagnosed with macrocephaly will receive further medical tests to determine whether the syndrome is accompanied by particular disorders. Those with benign or familial macrocephaly are considered to have megalencephaly.

Brachycephaly is the shape of a skull shorter than average in its species. It is perceived as a desirable trait in some domesticated dog and cat breeds, notably the pug and Persian, and can be normal or abnormal in other animal species.
Hereditary spastic paraplegia (HSP) is a group of inherited diseases whose main feature is a progressive gait disorder. The disease presents with progressive stiffness (spasticity) and contraction in the lower limbs. HSP is also known as hereditary spastic paraparesis, familial spastic paraplegia, French settlement disease, Strumpell disease, or Strumpell-Lorrain disease. The symptoms are a result of dysfunction of long axons in the spinal cord. The affected cells are the primary motor neurons; therefore, the disease is an upper motor neuron disease. HSP is not a form of cerebral palsy even though it physically may appear and behave much the same as spastic diplegia. The origin of HSP is different from cerebral palsy. Despite this, some of the same anti-spasticity medications used in spastic cerebral palsy are sometimes used to treat HSP symptoms.

Sabinas brittle hair syndrome, also called Sabinas syndrome or brittle hair-mental deficit syndrome, is an autosomal recessive congenital disorder affecting the integumentary system.

A unibrow is a single eyebrow created when the two eyebrows meet in the middle above the bridge of the nose. The hair above the bridge of the nose is of the same color and thickness as the eyebrows, such that they converge to form one uninterrupted line of hair.

Aniridia is the absence of the iris, a muscular structure that opens and closes the pupil to allow light into the eye. It is also responsible for eye color. Without it, the central eye appears all black. It can be congenital, in which both eyes are usually involved, or caused by a penetrant injury. Isolated aniridia is a congenital disorder that is not limited to a defect in iris development, but is a panocular condition with macular and optic nerve hypoplasia, cataract, and corneal changes. Vision may be severely compromised and the disorder is frequently associated with some ocular complications: nystagmus, amblyopia, buphthalmos, and cataract. Aniridia in some individuals occurs as part of a syndrome, such as WAGR syndrome, or Gillespie syndrome.
Short-chain acyl-coenzyme A dehydrogenase deficiency (SCADD) is an autosomal recessive fatty acid oxidation disorder which affects enzymes required to break down a certain group of fats called short chain fatty acids.

Laurence–Moon syndrome (LMS) is a rare autosomal recessive genetic disorder associated with retinitis pigmentosa, spastic paraplegia, and mental disabilities.

Sjögren–Larsson syndrome is a rare autosomal recessive form of ichthyosis with neurological symptoms. It can be identified by a triad of medical disorders. The first is ichthyosis, which is a buildup of skin to form a scale-like covering that causes dry skin and other problems. The second identifier is paraplegia which is characterized by leg spasms. The final identifier is intellectual delay.

Kaufman oculocerebrofacial syndrome, also known as blepharophimosis-ptosis-intellectual disability syndrome, is an extremely rare autosomal recessive congenital disorder characterized by severe mental retardation, brachycephaly, upslanting palpebral fissures, eye abnormalities, and highly arched palate. It was characterized in 1971; eight cases had been identified as of 1995. To date, the amount of cases is disputed, with sources claiming the number ranges from 14 to 31.
Congenital hemolytic anemia (CHA) is a diverse group of rare hereditary conditions marked by decreased life expectancy and premature removal of erythrocytes from blood flow. Defects in erythrocyte membrane proteins and red cell enzyme metabolism, as well as changes at the level of erythrocyte precursors, lead to impaired bone marrow erythropoiesis. CAH is distinguished by variable anemia, chronic extravascular hemolysis, decreased erythrocyte life span, splenomegaly, jaundice, biliary lithiasis, and iron overload. Immune-mediated mechanisms may play a role in the pathogenesis of these uncommon diseases, despite the paucity of data regarding the immune system's involvement in CHAs.
Kufor–Rakeb syndrome (KRS) is an autosomal recessive disorder of juvenile onset also known as Parkinson disease-9 (PARK9). It is named after Kufr Rakeb in Irbid, Jordan. Kufor–Rakeb syndrome was first identified in this region in Jordan with a Jordanian couple's 5 children who had rigidity, mask-like face, and bradykinesia. The disease was first described in 1994 by Najim Al-Din et al. The OMIM number is 606693.
Baller–Gerold syndrome (BGS) is a rare genetic syndrome that involves premature fusion of the skull bones and malformations of facial, forearm and hand bones. The symptoms of Baller–Gerold syndrome overlap with features of a few other genetics disorders: Rothmund–Thomson syndrome and RAPADILINO syndrome. The prevalence of BGS is unknown, as there have only been a few reported cases, but it is estimated to be less than 1 in a million. The name of the syndrome comes from the researchers Baller and Gerold who discovered the first three cases.
Barakat-Perenthaler syndrome is a rare neurodevelopmental genetic disorder, presenting with a severe epileptic encephalopathy, developmental delay, Intellectual disability, progressive microcephaly and visual disturbance. It is listed by the standard reference, Online Mendelian Inheritance in Man (OMIM) as #618744. and classified as EPILEPTIC ENCEPHALOPATHY, EARLY INFANTILE, 83; EIEE83. It was first described in 2019 by Dr. Stefan Barakat and his team at the Erasmus University Medical Center in Rotterdam in the journal Acta Neuropathologica; the most recent reviews were published in Epilepsy Currents. and Trends in Endocrinology and Metabolism
Spastic paraplegia 15 (SPG15) is a form of hereditary spastic paraplegia that commonly becomes apparent during childhood or adolescence. The disease is caused by mutations within the ZFYVE26 gene - also known as the SPG15 gene - and is passed down in an autosomal recessive manner.
Intellectual disability-spasticity-ectrodactyly syndrome, also known as Jancar syndrome, is a rare autosomal recessive genetic disorder which is characterized by severe intellectual disabilities, hereditary spastic paraplegia, and defects of the distal limbs, such as syndactyly, ectrodactyly, and clinodactyly. Only 3 families in England and Israel have been described in medical literature.

X-linked complicated corpus callosum dysgenesis is a genetic disorder characterized by dysplasia, hypoplasia or agenesis of the corpus callosum alongside variable intellectual disability and spastic paraplegia. Only 13 cases have been described in medical literature. Transmission is X-linked recessive. It is the mildest subtype of L1 syndrome.
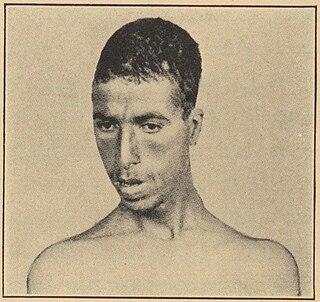
Narrow face is a condition in which the width of the face is abnormally reduced.

Low anterior hairline is a condition in which the frontal hairline which defines the top and sides of the forehead is unusually low. This can mean that either the distance between the trichion (hairline) and glabella at the midline is more than 2 SD below the mean, or that this distance is apparently (subjectively) decreased.
References
- ↑ Hereditary spastic paraplegia. PP Liberski, C Blackstone - Neurodegeneration, 2017
- ↑ Handley M, Sheridan E. RAB18 Deficiency. In: GeneReviews®. University of Washington, Seattle, Seattle (WA); 1993. PMID 29300443
- 1 2 3 4 5 "Warburg Micro Syndrome".
- ↑ Handley, Mark T.; et al. (2013). "Mutation Spectrum inRAB3GAP1,RAB3GAP2, andRAB18and Genotype-Phenotype Correlations in Warburg Micro Syndrome and Martsolf Syndrome". Human Mutation. 34 (5): 686–696. doi:10.1002/humu.22296. PMID 23420520. S2CID 2437070.
- ↑ "WARBURG Micro Syndrome." https://www.ncbi.nlm.nih.gov/ 10 Mar. 2008 <https://www.ncbi.nlm.nih.gov/omim/?term=600118>.
- ↑ Handley, Mark T.; Carpanini, Sarah M.; Mali, Girish R.; Sidjanin, Duska J.; Aligianis, Irene A.; Jackson, Ian J.; Fitzpatrick, David R. (2015). "Warburg Micro syndrome is caused by RAB18 deficiency or dysregulation". Open Biology. 5 (6): 150047. doi:10.1098/rsob.150047. PMC 4632505 . PMID 26063829.