Related Research Articles

Structural biology is a field that is many centuries old which, as defined by the Journal of Structural Biology, deals with structural analysis of living material at every level of organization. Early structural biologists throughout the 19th and early 20th centuries were primarily only able to study structures to the limit of the naked eye's visual acuity and through magnifying glasses and light microscopes.
The Protein Data Bank (PDB) is a database for the three-dimensional structural data of large biological molecules, such as proteins and nucleic acids. The data, typically obtained by X-ray crystallography, NMR spectroscopy, or, increasingly, cryo-electron microscopy, and submitted by biologists and biochemists from around the world, are freely accessible on the Internet via the websites of its member organisations. The PDB is overseen by an organization called the Worldwide Protein Data Bank, wwPDB.

A chemical structure of a molecule is a spatial arrangement of its atoms and their chemical bonds. Its determination includes a chemist's specifying the molecular geometry and, when feasible and necessary, the electronic structure of the target molecule or other solid. Molecular geometry refers to the spatial arrangement of atoms in a molecule and the chemical bonds that hold the atoms together and can be represented using structural formulae and by molecular models; complete electronic structure descriptions include specifying the occupation of a molecule's molecular orbitals. Structure determination can be applied to a range of targets from very simple molecules to very complex ones.

Structural bioinformatics is the branch of bioinformatics that is related to the analysis and prediction of the three-dimensional structure of biological macromolecules such as proteins, RNA, and DNA. It deals with generalizations about macromolecular 3D structures such as comparisons of overall folds and local motifs, principles of molecular folding, evolution, binding interactions, and structure/function relationships, working both from experimentally solved structures and from computational models. The term structural has the same meaning as in structural biology, and structural bioinformatics can be seen as a part of computational structural biology. The main objective of structural bioinformatics is the creation of new methods of analysing and manipulating biological macromolecular data in order to solve problems in biology and generate new knowledge.
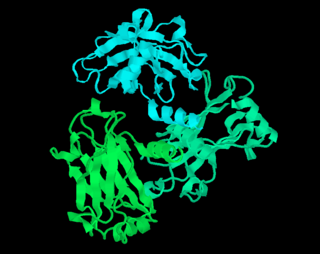
RasMol is a computer program written for molecular graphics visualization intended and used mainly to depict and explore biological macromolecule structures, such as those found in the Protein Data Bank. It was originally developed by Roger Sayle in the early 1990s.

Nuclear magnetic resonance spectroscopy, most commonly known as NMR spectroscopy or magnetic resonance spectroscopy (MRS), is a spectroscopic technique to observe local magnetic fields around atomic nuclei. This spectroscopy is based on the measurement of absorption of electromagnetic radiations in the radio frequency region from roughly 4 to 900 MHz. Absorption of radio waves in the presence of magnetic field is accompanied by a special type of nuclear transition, and for this reason, such type of spectroscopy is known as Nuclear Magnetic Resonance Spectroscopy. The sample is placed in a magnetic field and the NMR signal is produced by excitation of the nuclei sample with radio waves into nuclear magnetic resonance, which is detected with sensitive radio receivers. The intramolecular magnetic field around an atom in a molecule changes the resonance frequency, thus giving access to details of the electronic structure of a molecule and its individual functional groups. As the fields are unique or highly characteristic to individual compounds, in modern organic chemistry practice, NMR spectroscopy is the definitive method to identify monomolecular organic compounds.
Nuclear magnetic resonance spectroscopy of proteins is a field of structural biology in which NMR spectroscopy is used to obtain information about the structure and dynamics of proteins, and also nucleic acids, and their complexes. The field was pioneered by Richard R. Ernst and Kurt Wüthrich at the ETH, and by Ad Bax, Marius Clore, Angela Gronenborn at the NIH, and Gerhard Wagner at Harvard University, among others. Structure determination by NMR spectroscopy usually consists of several phases, each using a separate set of highly specialized techniques. The sample is prepared, measurements are made, interpretive approaches are applied, and a structure is calculated and validated.
Xplor-NIH is a highly sophisticated and flexible biomolecular structure determination program which includes an interface to the legacy X-PLOR program. The main developers are Charles Schwieters and Marius Clore of the National Institutes of Health. Xplor-NIH is based on a C++ framework with an extensive Python interface enabling very powerful and easy scripting of complex structure determination and refinement protocols. Restraints derived from all current solution and many solid state nuclear magnetic resonance (NMR) and X-ray scattering experiments can be accommodated during structure calculations. Extensive facilities are also available for many types of ensemble calculations where the experimental data cannot be accounted for by a unique structure. Many of the structure calculation protocols involve the use of simulated annealing designed to overcome local minima on the path of the global minimum region of the target function. These calculations can be carried out using any combination of Cartesian, torsion angle and rigid body dynamics and minimization. Currently Xplor-NIH is the most versatile, comprehensive and widely used structure determination/refinement package in NMR structure determination.
CNS or Crystallography and NMR system, is a software library for computational structural biology. It is an offshoot of X-PLOR and uses much of the same syntax. It is used in the fields of X-ray crystallography and NMR spectroscopy of biological macromolecules.
Molecular biophysics is a rapidly evolving interdisciplinary area of research that combines concepts in physics, chemistry, engineering, mathematics and biology. It seeks to understand biomolecular systems and explain biological function in terms of molecular structure, structural organization, and dynamic behaviour at various levels of complexity. This discipline covers topics such as the measurement of molecular forces, molecular associations, allosteric interactions, Brownian motion, and cable theory. Additional areas of study can be found on Outline of Biophysics. The discipline has required development of specialized equipment and procedures capable of imaging and manipulating minute living structures, as well as novel experimental approaches.
Carbohydrate NMR spectroscopy is the application of nuclear magnetic resonance (NMR) spectroscopy to structural and conformational analysis of carbohydrates. This method allows the scientists to elucidate structure of monosaccharides, oligosaccharides, polysaccharides, glycoconjugates and other carbohydrate derivatives from synthetic and natural sources. Among structural properties that could be determined by NMR are primary structure, saccharide conformation, stoichiometry of substituents, and ratio of individual saccharides in a mixture. Modern high field NMR instruments used for carbohydrate samples, typically 500 MHz or higher, are able to run a suite of 1D, 2D, and 3D experiments to determine a structure of carbohydrate compounds.

Nuclear magnetic resonance (NMR) is a physical phenomenon in which nuclei in a strong constant magnetic field are perturbed by a weak oscillating magnetic field and respond by producing an electromagnetic signal with a frequency characteristic of the magnetic field at the nucleus. This process occurs near resonance, when the oscillation frequency matches the intrinsic frequency of the nuclei, which depends on the strength of the static magnetic field, the chemical environment, and the magnetic properties of the isotope involved; in practical applications with static magnetic fields up to ca. 20 tesla, the frequency is similar to VHF and UHF television broadcasts (60–1000 MHz). NMR results from specific magnetic properties of certain atomic nuclei. Nuclear magnetic resonance spectroscopy is widely used to determine the structure of organic molecules in solution and study molecular physics and crystals as well as non-crystalline materials. NMR is also routinely used in advanced medical imaging techniques, such as in magnetic resonance imaging (MRI).
Nuclear magnetic resonance crystallography is a method which utilizes primarily NMR spectroscopy to determine the structure of solid materials on the atomic scale. Thus, solid-state NMR spectroscopy would be used primarily, possibly supplemented by quantum chemistry calculations, powder diffraction etc. If suitable crystals can be grown, any crystallographic method would generally be preferred to determine the crystal structure comprising in case of organic compounds the molecular structures and molecular packing. The main interest in NMR crystallography is in microcrystalline materials which are amenable to this method but not to X-ray, neutron and electron diffraction. This is largely because interactions of comparably short range are measured in NMR crystallography.
The following outline is provided as an overview of and topical guide to biophysics:
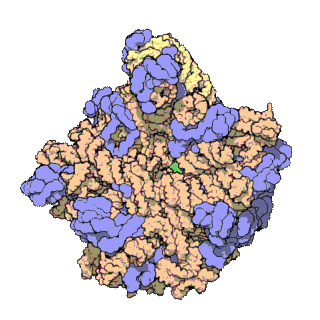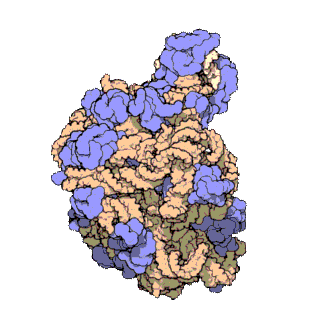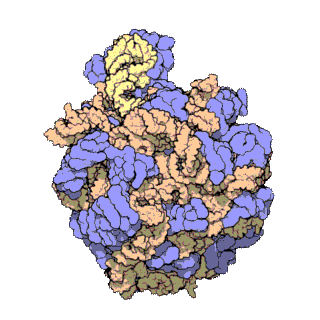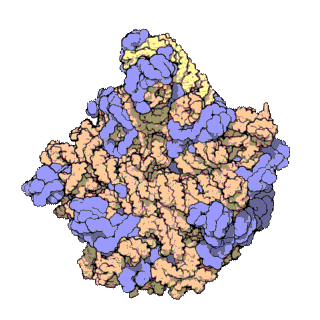
The term macromolecular assembly (MA) refers to massive chemical structures such as viruses and non-biologic nanoparticles, cellular organelles and membranes and ribosomes, etc. that are complex mixtures of polypeptide, polynucleotide, polysaccharide or other polymeric macromolecules. They are generally of more than one of these types, and the mixtures are defined spatially, and with regard to their underlying chemical composition and structure. Macromolecules are found in living and nonliving things, and are composed of many hundreds or thousands of atoms held together by covalent bonds; they are often characterized by repeating units. Assemblies of these can likewise be biologic or non-biologic, though the MA term is more commonly applied in biology, and the term supramolecular assembly is more often applied in non-biologic contexts. MAs of macromolecules are held in their defined forms by non-covalent intermolecular interactions, and can be in either non-repeating structures, or in repeating linear, circular, spiral, or other patterns. The process by which MAs are formed has been termed molecular self-assembly, a term especially applied in non-biologic contexts. A wide variety of physical/biophysical, chemical/biochemical, and computational methods exist for the study of MA; given the scale of MAs, efforts to elaborate their composition and structure and discern mechanisms underlying their functions are at the forefront of modern structure science.

Macromolecular structure validation is the process of evaluating reliability for 3-dimensional atomic models of large biological molecules such as proteins and nucleic acids. These models, which provide 3D coordinates for each atom in the molecule, come from structural biology experiments such as x-ray crystallography or nuclear magnetic resonance (NMR). The validation has three aspects: 1) checking on the validity of the thousands to millions of measurements in the experiment; 2) checking how consistent the atomic model is with those experimental data; and 3) checking consistency of the model with known physical and chemical properties.

In computational chemistry, conformational ensembles, also known as structural ensembles, are experimentally constrained computational models describing the structure of intrinsically unstructured proteins. Such proteins are flexible in nature, lacking a stable tertiary structure, and therefore cannot be described with a single structural representation. The techniques of ensemble calculation are relatively new on the field of structural biology, and are still facing certain limitations that need to be addressed before it will become comparable to classical structural description methods such as biological macromolecular crystallography.
Protein chemical shift prediction is a branch of biomolecular nuclear magnetic resonance spectroscopy that aims to accurately calculate protein chemical shifts from protein coordinates. Protein chemical shift prediction was first attempted in the late 1960s using semi-empirical methods applied to protein structures solved by X-ray crystallography. Since that time protein chemical shift prediction has evolved to employ much more sophisticated approaches including quantum mechanics, machine learning and empirically derived chemical shift hypersurfaces. The most recently developed methods exhibit remarkable precision and accuracy.

Gaetano T. Montelione is an American biophysical chemist, Professor of Chemistry and Chemical Biology, and Constellation Endowed Chair in Structural Bioinformatics at Rensselaer Polytechnic Institute in Troy, NY.
References
- ↑ Güntert, Peter (2011). "Automated protein structure determination from NMR data". In Dingley, Andrew J.; Pascal, Steven M. (eds.). Biomolecular NMR spectroscopy. Advances in Biomedical Spectroscopy. Vol. 3. Amsterdam: IOS Press. p. 341. doi:10.3233/978-1-60750-695-9-338. ISBN 9781607506942.