
In chemistry, an alkene is a hydrocarbon containing a carbon–carbon double bond.
The following outline is provided as an overview of and topical guide to organic chemistry:
An ylide or ylid is a neutral dipolar molecule containing a formally negatively charged atom (usually a carbanion) directly attached to a heteroatom with a formal positive charge (usually nitrogen, phosphorus or sulfur), and in which both atoms have full octets of electrons. The result can be viewed as a structure in which two adjacent atoms are connected by both a covalent and an ionic bond; normally written X+–Y−. Ylides are thus 1,2-dipolar compounds, and a subclass of zwitterions. They appear in organic chemistry as reagents or reactive intermediates.
In organic chemistry, a nucleophilic addition reaction is an addition reaction where a chemical compound with an electrophilic double or triple bond reacts with a nucleophile, such that the double or triple bond is broken. Nucleophilic additions differ from electrophilic additions in that the former reactions involve the group to which atoms are added accepting electron pairs, whereas the latter reactions involve the group donating electron pairs.
The Wolff–Kishner reduction is a reaction used in organic chemistry to convert carbonyl functionalities into methylene groups. In the context of complex molecule synthesis, it is most frequently employed to remove a carbonyl group after it has served its synthetic purpose of activating an intermediate in a preceding step. As such, there is no obvious retron for this reaction. The reaction was reported by Nikolai Kischner in 1911 and Ludwig Wolff in 1912,
The 1,3-dipolar cycloaddition is a chemical reaction between a 1,3-dipole and a dipolarophile to form a five-membered ring. The earliest 1,3-dipolar cycloadditions were described in the late 19th century to the early 20th century, following the discovery of 1,3-dipoles. Mechanistic investigation and synthetic application were established in the 1960s, primarily through the work of Rolf Huisgen. Hence, the reaction is sometimes referred to as the Huisgen cycloaddition. 1,3-dipolar cycloaddition is an important route to the regio- and stereoselective synthesis of five-membered heterocycles and their ring-opened acyclic derivatives. The dipolarophile is typically an alkene or alkyne, but can be other pi systems. When the dipolarophile is an alkyne, aromatic rings are generally produced.

The Simmons–Smith reaction is an organic cheletropic reaction involving an organozinc carbenoid that reacts with an alkene to form a cyclopropane. It is named after Howard Ensign Simmons, Jr. and Ronald D. Smith. It uses a methylene free radical intermediate that is delivered to both carbons of the alkene simultaneously, therefore the configuration of the double bond is preserved in the product and the reaction is stereospecific.
The Wittig reaction or Wittig olefination is a chemical reaction of an aldehyde or ketone with a triphenyl phosphonium ylide called a Wittig reagent. Wittig reactions are most commonly used to convert aldehydes and ketones to alkenes. Most often, the Wittig reaction is used to introduce a methylene group using methylenetriphenylphosphorane (Ph3P=CH2). Using this reagent, even a sterically hindered ketone such as camphor can be converted to its methylene derivative.
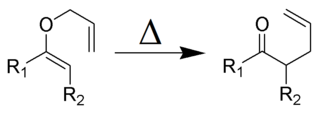
The Claisen rearrangement is a powerful carbon–carbon bond-forming chemical reaction discovered by Rainer Ludwig Claisen. The heating of an allyl vinyl ether will initiate a [3,3]-sigmatropic rearrangement to give a γ,δ-unsaturated carbonyl.
The Overman rearrangement is a chemical reaction that can be described as a Claisen rearrangement of allylic alcohols to give allylic trichloroacetamides through an imidate intermediate. The Overman rearrangement was discovered in 1974 by Larry Overman.

The Johnson–Corey–Chaykovsky reaction is a chemical reaction used in organic chemistry for the synthesis of epoxides, aziridines, and cyclopropanes. It was discovered in 1961 by A. William Johnson and developed significantly by E. J. Corey and Michael Chaykovsky. The reaction involves addition of a sulfur ylide to a ketone, aldehyde, imine, or enone to produce the corresponding 3-membered ring. The reaction is diastereoselective favoring trans substitution in the product regardless of the initial stereochemistry. The synthesis of epoxides via this method serves as an important retrosynthetic alternative to the traditional epoxidation reactions of olefins.

Azomethine ylides are nitrogen-based 1,3-dipoles, consisting of an iminium ion next to a carbanion. They are used in 1,3-dipolar cycloaddition reactions to form five-membered heterocycles, including pyrrolidines and pyrrolines. These reactions are highly stereo- and regioselective, and have the potential to form four new contiguous stereocenters. Azomethine ylides thus have high utility in total synthesis, and formation of chiral ligands and pharmaceuticals. Azomethine ylides can be generated from many sources, including aziridines, imines, and iminiums. They are often generated in situ, and immediately reacted with dipolarophiles.
The Curtin–Hammett principle is a principle in chemical kinetics proposed by David Yarrow Curtin and Louis Plack Hammett. It states that, for a reaction that has a pair of reactive intermediates or reactants that interconvert rapidly, each going irreversibly to a different product, the product ratio will depend both on the difference in energy between the two conformers and the energy barriers from each of the rapidly equilibrating isomers to their respective products. Stated another way, the product distribution reflects the difference in energy between the two rate-limiting transition states. As a result, the product distribution will not necessarily reflect the equilibrium distribution of the two intermediates. The Curtin–Hammett principle has been invoked to explain selectivity in a variety of stereo- and regioselective reactions. The relationship between the (apparent) rate constants and equilibrium constant is known as the Winstein-Holness equation.
In organic chemistry the Brook rearrangement refers to any [1,n] carbon to oxygen silyl migration. The rearrangement was first observed in the late 1950s by Canadian chemist Adrian Gibbs Brook (1924–2013), after which the reaction is named. These migrations can be promoted in a number of different ways, including thermally, photolytically or under basic/acidic conditions. In the forward direction, these silyl migrations produce silyl ethers as products which is driven by the stability of the oxygen-silicon bond.

Allylic strain in organic chemistry is a type of strain energy resulting from the interaction between a substituent on one end of an olefin with an allylic substituent on the other end. If the substituents are large enough in size, they can sterically interfere with each other such that one conformer is greatly favored over the other. Allyic strain was first recognized in the literature in 1965 by Johnson and Malhotra. The authors were investigating cyclohexane conformations including endocyclic and exocylic double bonds when they noticed certain conformations were disfavored due to the geometry constraints caused by the double bond. Organic chemists capitalize on the rigidity resulting from allylic strain for use in asymmetric reactions.
The Ei mechanism, also known as a thermal syn elimination or a pericyclic syn elimination, in organic chemistry is a special type of elimination reaction in which two vicinal substituents on an alkane framework leave simultaneously via a cyclic transition state to form an alkene in a syn elimination. This type of elimination is unique because it is thermally activated and does not require additional reagents unlike regular eliminations which require an acid or base, or would in many cases involve charged intermediates. This reaction mechanism is often found in pyrolysis.
The [2,3]-Wittig rearrangement is the transformation of an allylic ether into a homoallylic alcohol via a concerted, pericyclic process. Because the reaction is concerted, it exhibits a high degree of stereocontrol, and can be employed early in a synthetic route to establish stereochemistry. The Wittig rearrangement requires strongly basic conditions, however, as a carbanion intermediate is essential. [1,2]-Wittig rearrangement is a competitive process.
Desulfonylation reactions are chemical reactions leading to the removal of a sulfonyl group from organic compounds. As the sulfonyl functional group is electron-withdrawing, methods for cleaving the sulfur-carbon bonds of sulfones are typically reductive in nature. Olefination or replacement with hydrogen may be accomplished using reductive desulfonylation methods.
William Clark Still is an American organic chemist. As a distinguished professor at Columbia University, Clark Still made significant contributions to the field of organic chemistry, particularly in the areas of natural product synthesis, reaction development, conformational analysis, macrocyclic stereocontrol, and computational chemistry. Still and coworkers also developed the purification technique known as flash column chromatography which is widely used for the purification of organic compounds.
Rearrangements, especially those that can participate in cascade reactions, such as the aza-Cope rearrangements, are of high practical as well as conceptual importance in organic chemistry, due to their ability to quickly build structural complexity out of simple starting materials. The aza-Cope rearrangements are examples of heteroatom versions of the Cope rearrangement, which is a [3,3]-sigmatropic rearrangement that shifts single and double bonds between two allylic components. In accordance with the Woodward-Hoffman rules, thermal aza-Cope rearrangements proceed suprafacially. Aza-Cope rearrangements are generally classified by the position of the nitrogen in the molecule :



