454 Life Sciences was a biotechnology company based in Branford, Connecticut that specialized in high-throughput DNA sequencing. It was acquired by Roche in 2007 and shut down by Roche in 2013 when its technology became noncompetitive, although production continued until mid-2016.[1]
454 Life Sciences was founded by Jonathan Rothberg[2] and was originally known as 454 Corporation, a subsidiary of CuraGen. For their method for low-cost gene sequencing, 454 Life Sciences was awarded the Wall Street Journal's Gold Medal for Innovation in the Biotech-Medical category in 2005.[3] The name 454 was the code name by which the project was referred to at CuraGen, and the numbers have no known special meaning.[4]
In November 2006, Rothberg, Michael Egholm, and colleagues at 454 published a cover article with Svante Pääbo in Nature describing the first million base pairs of the Neanderthal genome, and initiated the Neanderthal Genome Project to complete the sequence of the Neanderthal genome by 2009.[5]
In late March 2007, Roche Diagnostics acquired 454 Life Sciences for US$154.9 million.[6] It remained a separate business unit.[7] In October 2013, Roche announced that it would shut down 454, and stop supporting the platform by mid-2016.[8]
In May 2007, 454 published the results of Project "Jim": the sequencing of the genome of James Watson, co-discoverer of the structure of DNA.[9][10]
Technology
454 Sequencing used a large-scale parallel pyrosequencing system capable of sequencing roughly 400-600 megabases of DNA per 10-hour run on the Genome Sequencer FLX with GS FLX Titanium series reagents.[11]
The system relied on fixing nebulized and adapter-ligated DNA fragments to small DNA-capture beads in a water-in-oil emulsion. The DNA fixed to these beads was then amplified by PCR. Each DNA-bound bead was placed into a ~29 μm well on a PicoTiterPlate, a fiber optic chip. A mix of enzymes such as DNA polymerase, ATP sulfurylase, and luciferase was also packed into the well. The PicoTiterPlate was then placed into the GS FLX System for sequencing.
454 released the GS20 sequencing machine in 2005, the first next-generation DNA sequencer on the market. In 2008, 454 Sequencing launched the GS FLX Titanium series reagents for use on the Genome Sequencer FLX instrument, with the ability to sequence 400-600 million base pairs per run with 400-500 base pair read lengths. In late 2009, 454 Life Sciences introduced the GS Junior System, a bench top version of the Genome Sequencer FLX System.[12]
DNA library preparation and emPCR
Genomic DNA was fractionated into smaller fragments (300-800 base pairs) and polished (made blunt at each end). Short adaptors were then ligated onto the ends of the fragments. These adaptors provided priming sequences for both amplification and sequencing of the sample-library fragments. One adaptor (Adaptor B) contained a 5'-biotin tag for immobilization of the DNA library onto streptavidin-coated beads. After nick repair, the non-biotinylated strand was released and used as a single-stranded template DNA (sstDNA) library. The sstDNA library was assessed for its quality, and the optimal amount (DNA copies per bead) needed for emPCR is determined by titration.[13]
The sstDNA library was immobilized onto beads. The beads containing a library fragment carried a single sstDNA molecule. The bead-bound library was emulsified with the amplification reagents in a water-in-oil mixture. Each bead was captured within its own microreactor where PCR amplification occurs. This resulted in bead-immobilized, clonally amplified DNA fragments.
Single-stranded template DNA library beads were added to the DNA Bead Incubation Mix (containing DNA polymerase) and were layered with Enzyme Beads (containing sulfurylase and luciferase) onto a PicoTiterPlate device. The device was centrifuged to deposit the beads into the wells. The layer of Enzyme Beads ensured that the DNA beads remained positioned in the wells during the sequencing reaction. The bead-deposition process was designed to maximize the number of wells that contain a single amplified library bead.
The loaded PicoTiterPlate device were placed into the Genome Sequencer FLX Instrument. The fluidics sub-system delivered sequencing reagents (containing buffers and nucleotides) across the wells of the plate. The four DNA nucleotides were added sequentially in a fixed order across the PicoTiterPlate device during a sequencing run. During the nucleotide flow, millions of copies of DNA bound to each of the beads were sequenced in parallel. When a nucleotide complementary to the template strand was added into a well, the polymerase extended the existing DNA strand by adding nucleotide(s). Addition of one (or more) nucleotide(s) generated a light signal that was recorded by the CCD camera in the instrument. This technique was based on sequencing-by-synthesis and called pyrosequencing.[14] The signal strength was proportional to the number of nucleotides; for example, homopolymer stretches, incorporated in a single nucleotide flow, generated a greater signal than single nucleotides. However, the signal strength for homopolymer stretches was linear only up to eight consecutive nucleotides, after which the signal fell off rapidly.[15] Data were stored in standard flowgram format (SFF) files for downstream analysis.
↑ Totty, Michael (October 24, 2005). "A Better Idea". The Wall Street Journal. Archived from the original on September 30, 2015. Retrieved March 13, 2017.
In genetics and biochemistry, sequencing means to determine the primary structure of an unbranched biopolymer. Sequencing results in a symbolic linear depiction known as a sequence which succinctly summarizes much of the atomic-level structure of the sequenced molecule.
A DNA sequencer is a scientific instrument used to automate the DNA sequencing process. Given a sample of DNA, a DNA sequencer is used to determine the order of the four bases: G (guanine), C (cytosine), A (adenine) and T (thymine). This is then reported as a text string, called a read. Some DNA sequencers can be also considered optical instruments as they analyze light signals originating from fluorochromes attached to nucleotides.
Pyrosequencing is a method of DNA sequencing based on the "sequencing by synthesis" principle, in which the sequencing is performed by detecting the nucleotide incorporated by a DNA polymerase. Pyrosequencing relies on light detection based on a chain reaction when pyrophosphate is released. Hence, the name pyrosequencing.
DNA sequencing is the process of determining the nucleic acid sequence – the order of nucleotides in DNA. It includes any method or technology that is used to determine the order of the four bases: adenine, guanine, cytosine, and thymine. The advent of rapid DNA sequencing methods has greatly accelerated biological and medical research and discovery.
Sanger sequencing is a method of DNA sequencing that involves electrophoresis and is based on the random incorporation of chain-terminating dideoxynucleotides by DNA polymerase during in vitro DNA replication. After first being developed by Frederick Sanger and colleagues in 1977, it became the most widely used sequencing method for approximately 40 years. It was first commercialized by Applied Biosystems in 1986. More recently, higher volume Sanger sequencing has been replaced by next generation sequencing methods, especially for large-scale, automated genome analyses. However, the Sanger method remains in wide use for smaller-scale projects and for validation of deep sequencing results. It still has the advantage over short-read sequencing technologies in that it can produce DNA sequence reads of > 500 nucleotides and maintains a very low error rate with accuracies around 99.99%. Sanger sequencing is still actively being used in efforts for public health initiatives such as sequencing the spike protein from SARS-CoV-2 as well as for the surveillance of norovirus outbreaks through the Center for Disease Control and Prevention's (CDC) CaliciNet surveillance network.
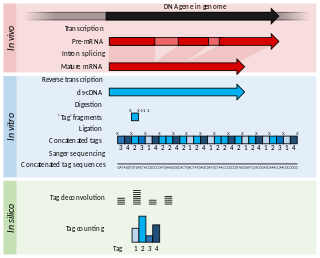
Serial Analysis of Gene Expression (SAGE) is a transcriptomic technique used by molecular biologists to produce a snapshot of the messenger RNA population in a sample of interest in the form of small tags that correspond to fragments of those transcripts. Several variants have been developed since, most notably a more robust version, LongSAGE, RL-SAGE and the most recent SuperSAGE. Many of these have improved the technique with the capture of longer tags, enabling more confident identification of a source gene.
The Neanderthal genome project is an effort of a group of scientists to sequence the Neanderthal genome, founded in July 2006.
Bisulfitesequencing (also known as bisulphite sequencing) is the use of bisulfite treatment of DNA before routine sequencing to determine the pattern of methylation. DNA methylation was the first discovered epigenetic mark, and remains the most studied. In animals it predominantly involves the addition of a methyl group to the carbon-5 position of cytosine residues of the dinucleotide CpG, and is implicated in repression of transcriptional activity.
Jonathan Marc Rothberg is an American scientist and entrepreneur. He is best known for his contributions to next-generation DNA sequencing. He works and resides in Guilford, Connecticut.
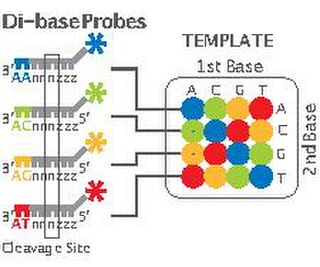
SOLiD (Sequencing by Oligonucleotide Ligation and Detection) is a next-generation DNA sequencing technology developed by Life Technologies and has been commercially available since 2006. This next generation technology generates 108 - 109 small sequence reads at one time. It uses 2 base encoding to decode the raw data generated by the sequencing platform into sequence data.
2 Base Encoding, also called SOLiD, is a next-generation sequencing technology developed by Applied Biosystems and has been commercially available since 2008. These technologies generate hundreds of thousands of small sequence reads at one time. Well-known examples of such DNA sequencing methods include 454 pyrosequencing, the Solexa system and the SOLiD system. These methods have reduced the cost from $0.01/base in 2004 to nearly $0.0001/base in 2006 and increased the sequencing capacity from 1,000,000 bases/machine/day in 2004 to more than 100,000,000 bases/machine/day in 2006.
Newbler is a software package for de novo DNA sequence assembly. It is designed specifically for assembling sequence data generated by the 454 GS-series of pyrosequencing platforms sold by 454 Life Sciences, a Roche Diagnostics company.
COLD-PCR is a modified polymerase chain reaction (PCR) protocol that enriches variant alleles from a mixture of wildtype and mutation-containing DNA. The ability to preferentially amplify and identify minority alleles and low-level somatic DNA mutations in the presence of excess wildtype alleles is useful for the detection of mutations. Detection of mutations is important in the case of early cancer detection from tissue biopsies and body fluids such as blood plasma or serum, assessment of residual disease after surgery or chemotherapy, disease staging and molecular profiling for prognosis or tailoring therapy to individual patients, and monitoring of therapy outcome and cancer remission or relapse. Common PCR will amplify both the major (wildtype) and minor (mutant) alleles with the same efficiency, occluding the ability to easily detect the presence of low-level mutations. The capacity to detect a mutation in a mixture of variant/wildtype DNA is valuable because this mixture of variant DNAs can occur when provided with a heterogeneous sample – as is often the case with cancer biopsies. Currently, traditional PCR is used in tandem with a number of different downstream assays for genotyping or the detection of somatic mutations. These can include the use of amplified DNA for RFLP analysis, MALDI-TOF genotyping, or direct sequencing for detection of mutations by Sanger sequencing or pyrosequencing. Replacing traditional PCR with COLD-PCR for these downstream assays will increase the reliability in detecting mutations from mixed samples, including tumors and body fluids.
Ion semiconductor sequencing is a method of DNA sequencing based on the detection of hydrogen ions that are released during the polymerization of DNA. This is a method of "sequencing by synthesis", during which a complementary strand is built based on the sequence of a template strand.
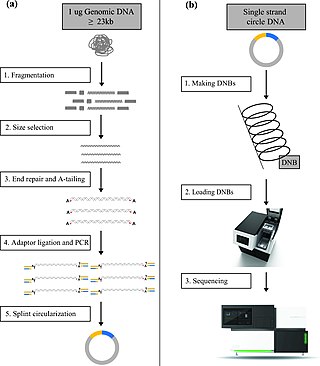
DNA nanoball sequencing is a high throughput sequencing technology that is used to determine the entire genomic sequence of an organism. The method uses rolling circle replication to amplify small fragments of genomic DNA into DNA nanoballs. Fluorescent nucleotides bind to complementary nucleotides and are then polymerized to anchor sequences bound to known sequences on the DNA template. The base order is determined via the fluorescence of the bound nucleotides This DNA sequencing method allows large numbers of DNA nanoballs to be sequenced per run at lower reagent costs compared to other next generation sequencing platforms. However, a limitation of this method is that it generates only short sequences of DNA, which presents challenges to mapping its reads to a reference genome. After purchasing Complete Genomics, the Beijing Genomics Institute (BGI) refined DNA nanoball sequencing to sequence nucleotide samples on their own platform.
Massive parallel sequencing or massively parallel sequencing is any of several high-throughput approaches to DNA sequencing using the concept of massively parallel processing; it is also called next-generation sequencing (NGS) or second-generation sequencing. Some of these technologies emerged between 1993 and 1998 and have been commercially available since 2005. These technologies use miniaturized and parallelized platforms for sequencing of 1 million to 43 billion short reads per instrument run.
Illumina dye sequencing is a technique used to determine the series of base pairs in DNA, also known as DNA sequencing. The reversible terminated chemistry concept was invented by Bruno Canard and Simon Sarfati at the Pasteur Institute in Paris. It was developed by Shankar Balasubramanian and David Klenerman of Cambridge University, who subsequently founded Solexa, a company later acquired by Illumina. This sequencing method is based on reversible dye-terminators that enable the identification of single nucleotides as they are washed over DNA strands. It can also be used for whole-genome and region sequencing, transcriptome analysis, metagenomics, small RNA discovery, methylation profiling, and genome-wide protein-nucleic acid interaction analysis.
Reduced representation bisulfite sequencing (RRBS) is an efficient and high-throughput technique for analyzing the genome-wide methylation profiles on a single nucleotide level. It combines restriction enzymes and bisulfite sequencing to enrich for areas of the genome with a high CpG content. Due to the high cost and depth of sequencing to analyze methylation status in the entire genome, Meissner et al. developed this technique in 2005 to reduce the amount of nucleotides required to sequence to 1% of the genome. The fragments that comprise the reduced genome still include the majority of promoters, as well as regions such as repeated sequences that are difficult to profile using conventional bisulfite sequencing approaches.
Paleogenomics is a field of science based on the reconstruction and analysis of genomic information in extinct species. Improved methods for the extraction of ancient DNA (aDNA) from museum artifacts, ice cores, archeological or paleontological sites, and next-generation sequencing technologies have spurred this field. It is now possible to detect genetic drift, ancient population migration and interrelationships, the evolutionary history of extinct plant, animal and Homo species, and identification of phenotypic features across geographic regions. Scientists can also use paleogenomics to compare ancient ancestors against modern-day humans. The rising importance of paleogenomics is evident from the fact that the 2022 Nobel Prize in physiology or medicine was awarded to a Swedish geneticist Svante Pääbo [1955-], who worked on paleogenomics.
BLESS, also known as breaks labeling, enrichment on streptavidin and next-generation sequencing, is a method used to detect genome-wide double-strand DNA damage. In contrast to chromatin immunoprecipitation (ChIP)-based methods of identifying DNA double-strand breaks (DSBs) by labeling DNA repair proteins, BLESS utilizes biotinylated DNA linkers to directly label genomic DNA in situ which allows for high-specificity enrichment of samples on streptavidin beads and the subsequent sequencing-based DSB mapping to nucleotide resolution.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.