Related Research Articles

Gene therapy is a medical technology that aims to produce a therapeutic effect through the manipulation of gene expression or through altering the biological properties of living cells.

Hearing loss is a partial or total inability to hear. Hearing loss may be present at birth or acquired at any time afterwards. Hearing loss may occur in one or both ears. In children, hearing problems can affect the ability to acquire spoken language, and in adults it can create difficulties with social interaction and at work. Hearing loss can be temporary or permanent. Hearing loss related to age usually affects both ears and is due to cochlear hair cell loss. In some people, particularly older people, hearing loss can result in loneliness.
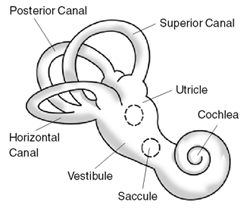
Ménière's disease (MD) is a disease of the inner ear that is characterized by potentially severe and incapacitating episodes of vertigo, tinnitus, hearing loss, and a feeling of fullness in the ear. Typically, only one ear is affected initially, but over time, both ears may become involved. Episodes generally last from 20 minutes to a few hours. The time between episodes varies. The hearing loss and ringing in the ears can become constant over time.
Tinnitus is a variety of sound that is heard when no corresponding external sound is present. Nearly everyone experiences faint "normal tinnitus" in a completely quiet room; but it is of concern only if it is bothersome, interferes with normal hearing, or is associated with other problems. The word tinnitus comes from the Latin tinnire, "to ring". In some people, it interferes with concentration, and can be associated with anxiety and depression.

Retinitis pigmentosa (RP) is a genetic disorder of the eyes that causes loss of vision. Symptoms include trouble seeing at night and decreasing peripheral vision. As peripheral vision worsens, people may experience "tunnel vision". Complete blindness is uncommon. Onset of symptoms is generally gradual and often begins in childhood.

Alport syndrome is a genetic disorder affecting around 1 in 5,000-10,000 children, characterized by glomerulonephritis, end-stage kidney disease, and hearing loss. Alport syndrome can also affect the eyes, though the changes do not usually affect vision, except when changes to the lens occur in later life. Blood in urine is universal. Proteinuria is a feature as kidney disease progresses.

Usher syndrome, also known as Hallgren syndrome, Usher–Hallgren syndrome, retinitis pigmentosa–dysacusis syndrome or dystrophia retinae dysacusis syndrome, is a rare genetic disorder caused by a mutation in any one of at least 11 genes resulting in a combination of hearing loss and visual impairment. It is a major cause of deafblindness and is at present incurable.
Friedreich's ataxia is an autosomal-recessive genetic disease that causes difficulty walking, a loss of coordination in the arms and legs, and impaired speech that worsens over time. Symptoms generally start between 5 and 20 years of age. Many develop hypertrophic cardiomyopathy and require a mobility aid such as a cane, walker, or wheelchair in their teens. As the disease progresses, some affected people lose their sight and hearing. Other complications may include scoliosis and diabetes mellitus.

In the inner ear, stereocilia are the mechanosensing organelles of hair cells, which respond to fluid motion in numerous types of animals for various functions, including hearing and balance. They are about 10–50 micrometers in length and share some similar features of microvilli. The hair cells turn the fluid pressure and other mechanical stimuli into electric stimuli via the many microvilli that make up stereocilia rods. Stereocilia exist in the auditory and vestibular systems.
Smith–Magenis Syndrome (SMS), also known as 17p- syndrome, is a microdeletion syndrome characterized by an abnormality in the short (p) arm of chromosome 17. It has features including intellectual disability, facial abnormalities, difficulty sleeping, and numerous behavioral problems such as self-harm. Smith–Magenis syndrome affects an estimated between 1 in 15,000 to 1 in 25,000 individuals.
Nonsyndromic deafness is hearing loss that is not associated with other signs and symptoms. In contrast, syndromic deafness involves hearing loss that occurs with abnormalities in other parts of the body. Nonsyndromic deafness constitutes 75% of all hearing loss cases, and an estimated 100 genes are thought to be linked to this condition. About 80% are linked to autosomal recessive inheritance, 15% to autosomal dominant inheritance, 1-3% through the X chromosome, and 0.5-1% are associated with mitochondrial inheritance.

Choroideremia is a rare, X-linked recessive form of hereditary retinal degeneration that affects roughly 1 in 50,000 males. The disease causes a gradual loss of vision, starting with childhood night blindness, followed by peripheral vision loss and progressing to loss of central vision later in life. Progression continues throughout the individual's life, but both the rate of change and the degree of visual loss are variable among those affected, even within the same family.
Refsum disease is an autosomal recessive neurological disease that results in the over-accumulation of phytanic acid in cells and tissues. It is one of several disorders named after Norwegian neurologist Sigvald Bernhard Refsum (1907–1991). Refsum disease typically is adolescent onset and is diagnosed by above average levels of phytanic acid. Humans obtain the necessary phytanic acid primarily through diet. It is still unclear what function phytanic acid plays physiologically in humans, but has been found to regulate fatty acid metabolism in the liver of mice.
Pendrin is an anion exchange protein that in humans is encoded by the SLC26A4 gene . Pendrin was initially identified as a sodium-independent chloride-iodide exchanger with subsequent studies showing that it also accepts formate and bicarbonate as substrates. Pendrin is similar to the Band 3 transport protein found in red blood cells. Pendrin is the protein which is mutated in Pendred syndrome, which is an autosomal recessive disorder characterized by sensorineural hearing loss, goiter and a partial organification problem detectable by a positive perchlorate test.

Harmonin is a protein that in humans is encoded by the USH1C gene. It is expressed in sensory cells of the inner ear and retina, where it plays a role in hearing, balance, and vision. Mutations at the USH1C locus cause Usher syndrome type 1c and nonsyndromic sensorineural deafness.

Potassium voltage-gated channel subfamily KQT member 4, also known as voltage-gated potassium channel subunit Kv7.4, is a protein that in humans is encoded by the KCNQ4 gene.
Alpha-tectorin is a protein that in humans is encoded by the TECTA gene.

Otoferlin is a protein that in humans is encoded by the OTOF gene. It is involved in vesicle membrane fusion, and mutations in the OTOF gene are associated with a genetic form of deafness.

Transmembrane channel-like protein 1 is a protein that in humans is encoded by the TMC1 gene. TMC1 contains six transmembrane domains with both the C and N termini on the endoplasmic side of the membrane, as well as a large loop between domains 4 and 5. This topology is similar to that of transient receptor potential channels (TRPs), a family of proteins involved in the perception of senses such as temperature, taste, pressure, and vision. TMC1 has been located in the post-natal mouse cochlea, and knockouts for TMC1 and TMC2 result in both auditory and vestibular deficits indicating TMC1 is a molecular part of auditory transduction.
DB-OTO is an experimental gene therapy for otoferlin-related hearing loss developed by Regeneron Pharmaceuticals. It is delivered via adeno-associated virus.
References
- ↑ Jiang, Luoying; Wang, Daqi; He, Yingzi; Shu, Yilai (April 2023). "Advances in gene therapy hold promise for treating hereditary hearing loss". Molecular Therapy. 31 (4): 934–950. doi: 10.1016/j.ymthe.2023.02.001 .
- ↑ Hickox, Ann E.; Valero, Michelle D.; McLaughlin, James T.; Robinson, Gregory S.; Wellman, Jennifer A.; McKenna, Michael J.; Sewell, William F.; Simons, Emmanuel J. (November 2021). "Genetic Medicine for Hearing Loss: OTOF as Exemplar". Journal of the American Academy of Audiology. 32 (10): 646–653. doi:10.1055/s-0041-1730410.