Microevolution is the change in allele frequencies that occurs over time within a population. This change is due to four different processes: mutation, selection, gene flow and genetic drift. This change happens over a relatively short amount of time compared to the changes termed macroevolution.
A genetic screen or mutagenesis screen is an experimental technique used to identify and select individuals who possess a phenotype of interest in a mutagenized population. Hence a genetic screen is a type of phenotypic screen. Genetic screens can provide important information on gene function as well as the molecular events that underlie a biological process or pathway. While genome projects have identified an extensive inventory of genes in many different organisms, genetic screens can provide valuable insight as to how those genes function.
Gene duplication is a major mechanism through which new genetic material is generated during molecular evolution. It can be defined as any duplication of a region of DNA that contains a gene. Gene duplications can arise as products of several types of errors in DNA replication and repair machinery as well as through fortuitous capture by selfish genetic elements. Common sources of gene duplications include ectopic recombination, retrotransposition event, aneuploidy, polyploidy, and replication slippage.
A quantitative trait locus (QTL) is a locus that correlates with variation of a quantitative trait in the phenotype of a population of organisms. QTLs are mapped by identifying which molecular markers correlate with an observed trait. This is often an early step in identifying the actual genes that cause the trait variation.

Comparative genomics is a branch of biological research that examines genome sequences across a spectrum of species, spanning from humans and mice to a diverse array of organisms from bacteria to chimpanzees. This large-scale holistic approach compares two or more genomes to discover the similarities and differences between the genomes and to study the biology of the individual genomes. Comparison of whole genome sequences provides a highly detailed view of how organisms are related to each other at the gene level. By comparing whole genome sequences, researchers gain insights into genetic relationships between organisms and study evolutionary changes. The major principle of comparative genomics is that common features of two organisms will often be encoded within the DNA that is evolutionarily conserved between them. Therefore, Comparative genomics provides a powerful tool for studying evolutionary changes among organisms, helping to identify genes that are conserved or common among species, as well as genes that give unique characteristics of each organism. Moreover, these studies can be performed at different levels of the genomes to obtain multiple perspectives about the organisms.

An inversion is a chromosome rearrangement in which a segment of a chromosome becomes inverted within its original position. An inversion occurs when a chromosome undergoes a two breaks within the chromosomal arm, and the segment between the two breaks inserts itself in the opposite direction in the same chromosome arm. The breakpoints of inversions often happen in regions of repetitive nucleotides, and the regions may be reused in other inversions. Chromosomal segments in inversions can be as small as 1 kilobases or as large as 100 megabases. The number of genes captured by an inversion can range from a handful of genes to hundreds of genes. Inversions can happen either through ectopic recombination between repetitive sequences, or through chromosomal breakage followed by non-homologous end joining.
Genetic architecture is the underlying genetic basis of a phenotypic trait and its variational properties. Phenotypic variation for quantitative traits is, at the most basic level, the result of the segregation of alleles at quantitative trait loci (QTL). Environmental factors and other external influences can also play a role in phenotypic variation. Genetic architecture is a broad term that can be described for any given individual based on information regarding gene and allele number, the distribution of allelic and mutational effects, and patterns of pleiotropy, dominance, and epistasis.
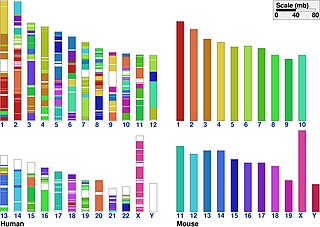
In genetics, the term synteny refers to two related concepts:
A polygene is a member of a group of non-epistatic genes that interact additively to influence a phenotypic trait, thus contributing to multiple-gene inheritance, a type of non-Mendelian inheritance, as opposed to single-gene inheritance, which is the core notion of Mendelian inheritance. The term "monozygous" is usually used to refer to a hypothetical gene as it is often difficult to distinguish the effect of an individual gene from the effects of other genes and the environment on a particular phenotype. Advances in statistical methodology and high throughput sequencing are, however, allowing researchers to locate candidate genes for the trait. In the case that such a gene is identified, it is referred to as a quantitative trait locus (QTL). These genes are generally pleiotropic as well. The genes that contribute to type 2 diabetes are thought to be mostly polygenes. In July 2016, scientists reported identifying a set of 355 genes from the last universal common ancestor (LUCA) of all organisms living on Earth.
Gene mapping or genome mapping describes the methods used to identify the location of a gene on a chromosome and the distances between genes. Gene mapping can also describe the distances between different sites within a gene.

Steven Dale Tanksley is the Chief Technology Officer of Nature Source Improved Plants. Prior to founding Nature Source Improved Plants, Tanksley served as the Liberty Hyde Bailey professor of plant breeding and biometry and chair of the Genomics Initiative Task Force at Cornell University College of Agriculture and Life Sciences. He is currently a Professor Emeritus at Cornell University.
Population genomics is the large-scale comparison of DNA sequences of populations. Population genomics is a neologism that is associated with population genetics. Population genomics studies genome-wide effects to improve our understanding of microevolution so that we may learn the phylogenetic history and demography of a population.
In genetics, association mapping, also known as "linkage disequilibrium mapping", is a method of mapping quantitative trait loci (QTLs) that takes advantage of historic linkage disequilibrium to link phenotypes to genotypes, uncovering genetic associations.
Nested association mapping (NAM) is a technique designed by the labs of Edward Buckler, James Holland, and Michael McMullen for identifying and dissecting the genetic architecture of complex traits in corn. It is important to note that nested association mapping is a specific technique that cannot be performed outside of a specifically designed population such as the Maize NAM population, the details of which are described below.
In statistical genetics, inclusive composite interval mapping (ICIM) has been proposed as an approach to QTL mapping for populations derived from bi-parental crosses. QTL mapping is based on genetic linkage map and phenotypic data to attempt to locate individual genetic factors on chromosomes and to estimate their genetic effects.
GeneNetwork is a combined database and open-source bioinformatics data analysis software resource for systems genetics. This resource is used to study gene regulatory networks that link DNA sequence differences to corresponding differences in gene and protein expression and to variation in traits such as health and disease risk. Data sets in GeneNetwork are typically made up of large collections of genotypes and phenotypes from groups of individuals, including humans, strains of mice and rats, and organisms as diverse as Drosophila melanogaster, Arabidopsis thaliana, and barley. The inclusion of genotypes makes it practical to carry out web-based gene mapping to discover those regions of genomes that contribute to differences among individuals in mRNA, protein, and metabolite levels, as well as differences in cell function, anatomy, physiology, and behavior.
A recombinant inbred strain or recombinant inbred line (RIL) is an organism with chromosomes that incorporate an essentially permanent set of recombination events between chromosomes inherited from two or more inbred strains. F1 and F2 generations are produced by intercrossing the inbred strains; pairs of the F2 progeny are then mated to establish inbred strains through long-term inbreeding.
Eviatar Nevo, is Professor Emeritus, founder and director of the Institute of Evolution at University of Haifa, Israel.

Peter D. Keightley is a British geneticist who is Professor of Evolutionary Genetics at the Institute of Evolutionary Biology in School of Biological Sciences at the University of Edinburgh.

Laboratory experiments of speciation have been conducted for all four modes of speciation: allopatric, peripatric, parapatric, and sympatric; and various other processes involving speciation: hybridization, reinforcement, founder effects, among others. Most of the experiments have been done on flies, in particular Drosophila fruit flies. However, more recent studies have tested yeasts, fungi, and even viruses.
