
Chiari malformation (CM) is a structural defect in the cerebellum, characterized by a downward displacement of one or both cerebellar tonsils through the foramen magnum. CMs can cause headaches, difficulty swallowing, vomiting, dizziness, neck pain, unsteady gait, poor hand coordination, numbness and tingling of the hands and feet, and speech problems. Less often, people may experience ringing or buzzing in the ears, weakness, slow heart rhythm, or fast heart rhythm, curvature of the spine (scoliosis) related to spinal cord impairment, abnormal breathing, such as central sleep apnea, characterized by periods of breathing cessation during sleep, and, in severe cases, paralysis.

Septo-optic dysplasia (SOD), known also as de Morsier syndrome, is a rare congenital malformation syndrome that features a combination of the underdevelopment of the optic nerve, pituitary gland dysfunction, and absence of the septum pellucidum . Two or more of these features need to be present for a clinical diagnosis—only 30% of patients have all three. French-Swiss doctor Georges de Morsier first recognized the relation of a rudimentary or absent septum pellucidum with hypoplasia of the optic nerves and chiasm in 1956.
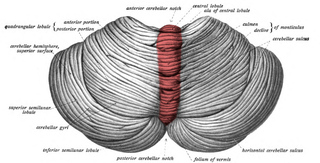
The cerebellar vermis is located in the medial, cortico-nuclear zone of the cerebellum, which is in the posterior fossa of the cranium. The primary fissure in the vermis curves ventrolaterally to the superior surface of the cerebellum, dividing it into anterior and posterior lobes. Functionally, the vermis is associated with bodily posture and locomotion. The vermis is included within the spinocerebellum and receives somatic sensory input from the head and proximal body parts via ascending spinal pathways.

Dandy–Walker malformation (DWM), also known as Dandy–Walker syndrome (DWS), is a rare congenital brain malformation in which the part joining the two hemispheres of the cerebellum does not fully form, and the fourth ventricle and space behind the cerebellum are enlarged with cerebrospinal fluid. Most of those affected develop hydrocephalus within the first year of life, which can present as increasing head size, vomiting, excessive sleepiness, irritability, downward deviation of the eyes and seizures. Other, less common symptoms are generally associated with comorbid genetic conditions and can include congenital heart defects, eye abnormalities, intellectual disability, congenital tumours, other brain defects such as agenesis of the corpus callosum, skeletal abnormalities, an occipital encephalocele or underdeveloped genitalia or kidneys. It is sometimes discovered in adolescents or adults due to mental health problems.

Walker–Warburg syndrome (WWS), also called Warburg syndrome, Chemke syndrome, HARD syndrome, Pagon syndrome, cerebroocular dysgenesis (COD) or cerebroocular dysplasia-muscular dystrophy syndrome (COD-MD), is a rare form of autosomal recessive congenital muscular dystrophy. It is associated with brain and eye abnormalities. This condition has a worldwide distribution. Walker-Warburg syndrome is estimated to affect 1 in 60,500 newborns worldwide.

Otocephaly, also known as agnathia–otocephaly complex, is a very rare and lethal cephalic disorder characterized by the absence of the mandible (agnathia), with the ears fused together just below the chin (synotia). It is caused by a disruption to the development of the first branchial arch. It occurs in every 1 in 70,000 embryos.

Spondylocostal dysostosis, also known as Jarcho-Levin syndrome (JLS), is a rare, heritable axial skeleton growth disorder. It is characterized by widespread and sometimes severe malformations of the vertebral column and ribs, shortened thorax, and moderate to severe scoliosis and kyphosis. Individuals with Jarcho-Levin typically appear to have a short trunk and neck, with arms appearing relatively long in comparison, and a slightly protuberant abdomen. Severely affected individuals may have life-threatening pulmonary complications due to deformities of the thorax. The syndrome was first described by Saul Jarcho and Paul M. Levin at Johns Hopkins University in 1938.
Ohtahara syndrome (OS), also known as early infantile epileptic encephalopathy (EIEE) is a progressive epileptic encephalopathy. The syndrome is outwardly characterized by tonic spasms and partial seizures within the first few months of life, and receives its more elaborate name from the pattern of burst activity on an electroencephalogram (EEG). It is an extremely debilitating progressive neurological disorder, involving intractable seizures and severe intellectual disabilities. No single cause has been identified, although in many cases structural brain damage is present.

Gillespie syndrome, also called aniridia, cerebellar ataxia and mental deficiency, is a rare genetic disorder. The disorder is characterized by partial aniridia, ataxia, and, in most cases, intellectual disability. It is heterogeneous, inherited in either an autosomal dominant or autosomal recessive manner. Gillespie syndrome was first described by American ophthalmologist Fredrick Gillespie in 1965.
XK aprosencephaly is an extremely rare congenital disorder characterized by the absence of the embryonic forebrain. Because the prosencephalon gives way to the cerebral cortex, survival with aprosencephaly is not possible outside utero. The external symptoms are similar to holoprosencephaly, a related disorder, including a smaller than normal head (microcephaly), small eyeballs (microphthalmia), a small mouth (microstomia), anal atresia, and abnormalities of the external genitalia, radius, nostrils, and pharynx (throat).
Cousin syndrome is a genetic condition characterized by short stature at birth, a short neck with low-positioned external ears, as well as congenital malformations of the skeletal system affecting the shoulders, the pelvis, the neck, and the limbs. The condition determines physical disability, particularly affecting deambulation, and hearing loss while intelligence is not affected.

A bifid nose is an uncommon congenital malformation which is characterized by the presence of a cleft between the two nostrils of the nose. It is the result of a disturbance during embryological nose development.

Chudley–Mccullough syndrome is a rare genetic disorder which is characterized by bilateral congenital hearing loss associated with brain malformations. It is a type of syndromic deafness.
Absence deformity of leg-cataract syndrome is a very rare genetic limb malformation which is characterized by unilateral absence deformity, scoliosis, low stature, and optic nerve dysplasia-associated congenital cataract. It has been described in two distantly related kin of Amish descent.
Infantile cerebral and cerebellar atrophy with postnatal progressive microcephaly is a rare hereditary autosomal recessive malformation syndrome of the central nervous system characterized by profound motor delays and intellectual disabilities, progressive microcephaly, hypertonia, spasticity, clonus and epilepsy. MRI findings include severe cerebellar and cerebral deterioration (atrophy) and impaired myelination. This condition is an example of consequences from the Founder effect, especially that of Jewish populations.

Wormian bone-multiple fractures-dentinogenesis imperfecta-skeletal dysplasia syndrome is a rare genetic bone disorder which is characterized by the presence of wormian bones in the skull, dentinogenesis imperfecta, recurrent bone fractures, hypertelorism, and eye puffiness. This disorder is unique from osteogenesis imperfecta because of the presence of cortical defects and the absence of defective collagen or osteopenia. It is not exactly known whether this condition is autosomal dominant or autosomal recessive.

X-linked complicated corpus callosum dysgenesis is a genetic disorder characterized by dysplasia, hypoplasia or agenesis of the corpus callosum alongside variable intellectual disability and spastic paraplegia. Only 13 cases have been described in medical literature. Transmission is X-linked recessive. It is the mildest subtype of L1 syndrome.
Porencephaly-cerebellar hypoplasia-internal malformations syndrome is a rare autosomal recessive syndrome that mainly affects the central nervous system. It causes cardiac defects, brain anomalies, and craniofacial dysmorphisms. It has been reported in a pair of German siblings of the opposite sex born to consanguineous Turkish parents.

Craniosynostosis-Dandy-Walker malformation-hydrocephalus syndrome is an autosomal dominant syndrome characterized by sagittal craniosynostosis (scaphocephaly), Dandy-Walker malformation, hydrocephalus, and craniofacial dysmorphisms including hypertelorism, micrognathia, and positional ear deformities.
