The ene reaction is a chemical reaction between an alkene with an allylic hydrogen and a compound containing a multiple bond, in order to form a new σ-bond with migration of the ene double bond and 1,5 hydrogen shift. The product is a substituted alkene with the double bond shifted to the allylic position.

BINAP (2,2′-bis(diphenylphosphino)-1,1′-binaphthyl) is an organophosphorus compound. This chiral diphosphine ligand is widely used in asymmetric synthesis. It consists of a pair of 2-diphenylphosphinonaphthyl groups linked at the 1 and 1′ positions. This C2-symmetric framework lacks a stereogenic atom, but has axial chirality due to restricted rotation (atropisomerism). The barrier to racemization is high due to steric hindrance, which limits rotation about the bond linking the naphthyl rings. The dihedral angle between the naphthyl groups is approximately 90°. The natural bite angle is 93°.
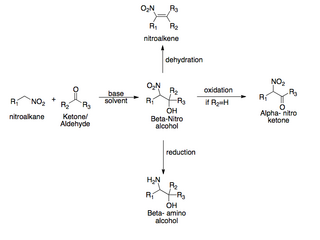
The Henry Reaction is a classic carbon–carbon bond formation reaction in organic chemistry. Discovered in 1895 by the Belgian chemist Louis Henry (1834-1913), it is the combination of a nitroalkane and an aldehyde or ketone in the presence of a base to form β-nitro alcohols. This type of reaction is commonly referred to as a "nitro-aldol" reaction. It is nearly analogous to the aldol reaction that had been discovered 23 years prior that couples two carbonyl compounds to form β-hydroxy carbonyl compounds known as "aldols". The Henry reaction is a useful technique in the area of organic chemistry due to the synthetic utility of its corresponding products, as they can be easily converted to other useful synthetic intermediates. These conversions include subsequent dehydration to yield nitroalkenes, oxidation of the secondary alcohol to yield α-nitro ketones, or reduction of the nitro group to yield β-amino alcohols.

The CBS catalyst or Corey–Bakshi–Shibata catalyst is an asymmetric catalyst derived from proline. It finds many uses in organic reactions such as the CBS reduction, Diels-Alder reactions and (3+2) cycloadditions. Proline, a naturally occurring chiral compound, is readily and cheaply available. It transfers its stereocenter to the catalyst which in turn is able to drive an organic reaction enantioselectively to one of two possible enantiomers. This selectivity is due to steric strain in the transition state that develops for one enantiomer but not for the other.
The Stetter reaction is a reaction used in organic chemistry to form carbon-carbon bonds through a 1,4-addition reaction utilizing a nucleophilic catalyst. While the related 1,2-addition reaction, the benzoin condensation, was known since the 1830s, the Stetter reaction was not reported until 1973 by Dr. Hermann Stetter. The reaction provides synthetically useful 1,4-dicarbonyl compounds and related derivatives from aldehydes and Michael acceptors. Unlike 1,3-dicarbonyls, which are easily accessed through the Claisen condensation, or 1,5-dicarbonyls, which are commonly made using a Michael reaction, 1,4-dicarbonyls are challenging substrates to synthesize, yet are valuable starting materials for several organic transformations, including the Paal–Knorr synthesis of furans and pyrroles. Traditionally utilized catalysts for the Stetter reaction are thiazolium salts and cyanide anion, but more recent work toward the asymmetric Stetter reaction has found triazolium salts to be effective. The Stetter reaction is an example of umpolung chemistry, as the inherent polarity of the aldehyde is reversed by the addition of the catalyst to the aldehyde, rendering the carbon center nucleophilic rather than electrophilic.
Transfer hydrogenation is the addition of hydrogen (H2; dihydrogen in inorganic and organometallic chemistry) to a molecule from a source other than gaseous H2. It is applied in industry and in organic synthesis, in part because of the inconvenience and expense of using gaseous H2. One large scale application of transfer hydrogenation is coal liquefaction using "donor solvents" such as tetralin.
The Meerwein–Ponndorf–Verley (MPV) reduction in organic chemistry is the reduction of ketones and aldehydes to their corresponding alcohols utilizing aluminium alkoxide catalysis in the presence of a sacrificial alcohol. The advantages of the MPV reduction lie in its high chemoselectivity, and its use of a cheap environmentally friendly metal catalyst.
In organic chemistry, kinetic resolution is a means of differentiating two enantiomers in a racemic mixture. In kinetic resolution, two enantiomers react with different reaction rates in a chemical reaction with a chiral catalyst or reagent, resulting in an enantioenriched sample of the less reactive enantiomer. As opposed to chiral resolution, kinetic resolution does not rely on different physical properties of diastereomeric products, but rather on the different chemical properties of the racemic starting materials. This enantiomeric excess (ee) of the unreacted starting material continually rises as more product is formed, reaching 100% just before full completion of the reaction. Kinetic resolution relies upon differences in reactivity between enantiomers or enantiomeric complexes. Kinetic resolution is a concept in organic chemistry and can be used for the preparation of chiral molecules in organic synthesis. Kinetic resolution reactions utilizing purely synthetic reagents and catalysts are much less common than the use of enzymatic kinetic resolution in application towards organic synthesis, although a number of useful synthetic techniques have been developed in the past 30 years.

Organozinc compounds in organic chemistry contain carbon to zinc chemical bonds. Organozinc chemistry is the science of organozinc compounds describing their physical properties, synthesis and reactions.
In chemistry, the Noyori asymmetric hydrogenation of ketones is a chemical reaction for the enantioselective hydrogenation of ketones, aldehydes, and imines. This reaction exploits using chiral ruthenium catalysts introduced by Ryoji Noyori. He shared half of the Nobel Prize in Chemistry in 2001 with William S. Knowles for the study of the asymmetric hydrogenation.

In organic chemistry, the term organocatalysis refers to a form of catalysis, whereby the rate of a chemical reaction is increased by an organic catalyst referred to as an "organocatalyst" consisting of carbon, hydrogen, sulfur and other nonmetal elements found in organic compounds. Because of their similarity in composition and description, they are often mistaken as a misnomer for enzymes due to their comparable effects on reaction rates and forms of catalysis involved.
Chiral Lewis acids (CLAs) are a type of Lewis acid catalyst that effects the chirality of the substrate as it reacts with it. In such reactions the synthesis favors the formation of a specific enantiomer or diastereomer. The method then is an enantioselective asymmetric synthesis reaction. Since they affect chirality, they produce optically active products from optically inactive or mixed starting materials. This type of preferential formation of one enantiomer or diastereomer over the other is formally known as an asymmetric induction. In this kind of Lewis acid. the electron-accepting atom is typically a metal, such as indium, zinc, lithium, aluminium, titanium, or boron. The chiral-altering ligands employed for synthesizing these acids most often have multiple Lewis basic sites that allow the formation of a ring structure involving the metal atom.
Nucleophilic epoxidation is the formation of epoxides from electron-deficient double bonds through the action of nucleophilic oxidants. Nucleophilic epoxidation methods represent a viable alternative to electrophilic methods, many of which do not epoxidize electron-poor double bonds efficiently.
Enantioselective ketone reductions convert prochiral ketones into chiral, non-racemic alcohols and are used heavily for the synthesis of stereodefined alcohols.
The Baylis–Hillman reaction is a carbon-carbon bond forming reaction between the α-position of an activated alkene and a carbon electrophile such as an aldehyde. Employing a nucleophilic catalyst, such as a tertiary amine and phosphine, this reaction provides a densely functionalized product. It is named for Anthony B. Baylis and Melville E. D. Hillman, two of the chemists who developed this reaction while working at Celanese. This reaction is also known as the Morita–Baylis–Hillman reaction or MBH reaction, as Morita had published earlier work on it.

Dynamic kinetic resolution in chemistry is a type of kinetic resolution where 100% of a racemic compound can be converted into a enantiopure compound. It is applied in asymmetric synthesis. Asymmetric synthesis has become a much explored field due to the challenge of creating a compound with a single 3D structure. Even more challenging is the ability to take a racemic mixture and have only one chiral product left after a reaction. One method that has become an exceedingly useful tool is dynamic kinetic resolution (DKR). DKR utilizes a center of a particular molecule that can be easily epimerized so that the (R) and (S) enantiomers can interconvert throughout the reaction process. At this point the catalyst can selectively lower the transition state energy of a single enantiomer, leading to almost 100% yield of one reaction pathway over the other. The figure below is an example of an energy diagram for a compound with an (R) and (S) isomer.

Asymmetric addition of alkenylmetals to aldehydes is a chemical reaction in enantioselective synthesis that reacts an alkenylmetal with an aldehyde to give an allyl alcohol. The stereoselectivity in the reaction is typically controlled by the asymmetric ligands used providing a strategy to introduce controlled asymmetry into the molecule. Controlled molecular asymmetry is crucial for controlling the bioactivity of the synthesized molecules and demanded by drug authorities in drug synthesis. In this case the ligands chelate to the transition metal to create a chiral environment which enables the selective formation of a particular enantiomer. Various transition metals such as Zinc, Nickel, Chromium, and Rhodium have been used in this reaction.
The asymmetric addition of alkynylzinc compounds to aldehydes is an example of a Nef synthesis, a chemical reaction whereby a chiral propargyl alcohol is prepared from a terminal alkyne and an aldehyde. This alkynylation reaction is enantioselective and involves an alkynylzinc reagent rather than the sodium acetylide used by John Ulric Nef in his 1899 report of the synthetic approach. Propargyl alcohols are versatile precursors for the chirally-selective synthesis of natural products and pharmaceutical agents, making this asymmetric addition reaction of alkynylzinc compounds useful. For example, Erick Carreira used this approach in a total synthesis of the marine natural product leucascandrolide A, a bioactive metabolite of the calcareous sponge Leucascandra caveolata with cytotoxic and antifungal properties isolated in 1996.

A phosphoramidite ligand is a chiral monodentate phosphine ligand, widely used for enantioselective synthesis. They were invented by Dutch chemist Ben Feringa. The introduction of phosphoramidite ligands challenged the notion that high flexibility in the metal–ligand complex is detrimental for high stereo control.
In homogeneous catalysis, a C2-symmetric ligands usually describes bidentate ligands that are dyssymmetric but not asymmetric by virtue of their C2-symmetry. Such ligands have proven valuable in catalysis. With C2 symmetry, C2-symmetric ligands limit the number of possible reaction pathways and thereby increase enantioselectivity, at least relative to asymmetrical analogues. Most chiral ligands combine with metals to form chiral catalyst engages in a chemical reaction in which chirality is transfer to the reaction product.