Related Research Articles

The polymerase chain reaction (PCR) is a method widely used to make millions to billions of copies of a specific DNA sample rapidly, allowing scientists to amplify a very small sample of DNA sufficiently to enable detailed study. PCR was invented in 1983 by American biochemist Kary Mullis at Cetus Corporation. Mullis and biochemist Michael Smith, who had developed other essential ways of manipulating DNA, were jointly awarded the Nobel Prize in Chemistry in 1993.

Reverse transcription polymerase chain reaction (RT-PCR) is a laboratory technique combining reverse transcription of RNA into DNA and amplification of specific DNA targets using polymerase chain reaction (PCR). It is primarily used to measure the amount of a specific RNA. This is achieved by monitoring the amplification reaction using fluorescence, a technique called real-time PCR or quantitative PCR (qPCR). Combined RT-PCR and qPCR are routinely used for analysis of gene expression and quantification of viral RNA in research and clinical settings.

DNA sequencing is the process of determining the nucleic acid sequence – the order of nucleotides in DNA. It includes any method or technology that is used to determine the order of the four bases: adenine, guanine, cytosine, and thymine. The advent of rapid DNA sequencing methods has greatly accelerated biological and medical research and discovery.
Cycling probe technology (CPT) is a molecular biological technique for detecting specific DNA sequences. CPT operates under isothermal conditions. In some applications, CPT offers an alternative to PCR. However, unlike PCR, CPT does not generate multiple copies of the target DNA itself, and the amplification of the signal is linear, in contrast to the exponential amplification of the target DNA in PCR. CPT uses a sequence specific chimeric probe which hybridizes to a complementary target DNA sequence and becomes a substrate for RNase H. Cleavage occurs at the RNA internucleotide linkages and results in dissociation of the probe from the target, thereby making it available for the next probe molecule. Integrated electrokinetic systems have been developed for use in CPT.

Bert Vogelstein is director of the Ludwig Center, Clayton Professor of Oncology and Pathology and a Howard Hughes Medical Institute investigator at The Johns Hopkins Medical School and Sidney Kimmel Comprehensive Cancer Center. A pioneer in the field of cancer genomics, his studies on colorectal cancers revealed that they result from the sequential accumulation of mutations in oncogenes and tumor suppressor genes. These studies now form the paradigm for modern cancer research and provided the basis for the notion of the somatic evolution of cancer.
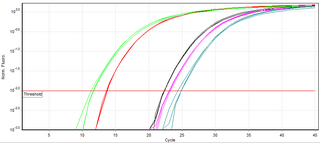
A real-time polymerase chain reaction is a laboratory technique of molecular biology based on the polymerase chain reaction (PCR). It monitors the amplification of a targeted DNA molecule during the PCR, not at its end, as in conventional PCR. Real-time PCR can be used quantitatively and semi-quantitatively.
SNP genotyping is the measurement of genetic variations of single nucleotide polymorphisms (SNPs) between members of a species. It is a form of genotyping, which is the measurement of more general genetic variation. SNPs are one of the most common types of genetic variation. An SNP is a single base pair mutation at a specific locus, usually consisting of two alleles. SNPs are found to be involved in the etiology of many human diseases and are becoming of particular interest in pharmacogenetics. Because SNPs are conserved during evolution, they have been proposed as markers for use in quantitative trait loci (QTL) analysis and in association studies in place of microsatellites. The use of SNPs is being extended in the HapMap project, which aims to provide the minimal set of SNPs needed to genotype the human genome. SNPs can also provide a genetic fingerprint for use in identity testing. The increase of interest in SNPs has been reflected by the furious development of a diverse range of SNP genotyping methods.
Multiplex ligation-dependent probe amplification (MLPA) is a variation of the multiplex polymerase chain reaction that permits amplification of multiple targets with only a single primer pair. It detects copy number changes at the molecular level, and software programs are used for analysis. Identification of deletions or duplications can indicate pathogenic mutations, thus MLPA is an important diagnostic tool used in clinical pathology laboratories worldwide.
Digital polymerase chain reaction is a biotechnological refinement of conventional polymerase chain reaction methods that can be used to directly quantify and clonally amplify nucleic acids strands including DNA, cDNA, or RNA. The key difference between dPCR and traditional PCR lies in the method of measuring nucleic acids amounts, with the former being a more precise method than PCR, though also more prone to error in the hands of inexperienced users. A "digital" measurement quantitatively and discretely measures a certain variable, whereas an “analog” measurement extrapolates certain measurements based on measured patterns. PCR carries out one reaction per single sample. dPCR also carries out a single reaction within a sample, however the sample is separated into a large number of partitions and the reaction is carried out in each partition individually. This separation allows a more reliable collection and sensitive measurement of nucleic acid amounts. The method has been demonstrated as useful for studying variations in gene sequences — such as copy number variants and point mutations — and it is routinely used for clonal amplification of samples for next-generation sequencing.
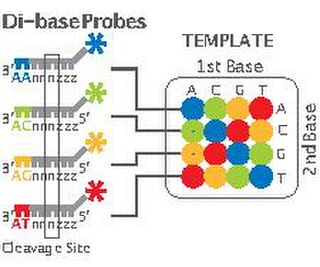
2 Base Encoding, also called SOLiD, is a next-generation sequencing technology developed by Applied Biosystems and has been commercially available since 2008. These technologies generate hundreds of thousands of small sequence reads at one time. Well-known examples of such DNA sequencing methods include 454 pyrosequencing, the Solexa system and the SOLiD system. These methods have reduced the cost from $0.01/base in 2004 to nearly $0.0001/base in 2006 and increased the sequencing capacity from 1,000,000 bases/machine/day in 2004 to more than 100,000,000 bases/machine/day in 2006.
In vitro compartmentalization (IVC) is an emulsion-based technology that generates cell-like compartments in vitro. These compartments are designed such that each contains no more than one gene. When the gene is transcribed and/or translated, its products become 'trapped' with the encoding gene inside the compartment. By coupling the genotype (DNA) and phenotype, compartmentalization allows the selection and evolution of phenotype.
Polony sequencing is an inexpensive but highly accurate multiplex sequencing technique that can be used to “read” millions of immobilized DNA sequences in parallel. This technique was first developed by Dr. George Church's group at Harvard Medical School. Unlike other sequencing techniques, Polony sequencing technology is an open platform with freely downloadable, open source software and protocols. Also, the hardware of this technique can be easily set up with a commonly available epifluorescence microscopy and a computer-controlled flowcell/fluidics system. Polony sequencing is generally performed on paired-end tags library that each molecule of DNA template is of 135 bp in length with two 17–18 bp paired genomic tags separated and flanked by common sequences. The current read length of this technique is 26 bases per amplicon and 13 bases per tag, leaving a gap of 4–5 bases in each tag.
COLD-PCR is a modified polymerase chain reaction (PCR) protocol that enriches variant alleles from a mixture of wildtype and mutation-containing DNA. The ability to preferentially amplify and identify minority alleles and low-level somatic DNA mutations in the presence of excess wildtype alleles is useful for the detection of mutations. Detection of mutations is important in the case of early cancer detection from tissue biopsies and body fluids such as blood plasma or serum, assessment of residual disease after surgery or chemotherapy, disease staging and molecular profiling for prognosis or tailoring therapy to individual patients, and monitoring of therapy outcome and cancer remission or relapse. Common PCR will amplify both the major (wildtype) and minor (mutant) alleles with the same efficiency, occluding the ability to easily detect the presence of low-level mutations. The capacity to detect a mutation in a mixture of variant/wildtype DNA is valuable because this mixture of variant DNAs can occur when provided with a heterogeneous sample – as is often the case with cancer biopsies. Currently, traditional PCR is used in tandem with a number of different downstream assays for genotyping or the detection of somatic mutations. These can include the use of amplified DNA for RFLP analysis, MALDI-TOF genotyping, or direct sequencing for detection of mutations by Sanger sequencing or pyrosequencing. Replacing traditional PCR with COLD-PCR for these downstream assays will increase the reliability in detecting mutations from mixed samples, including tumors and body fluids.
Massive parallel sequencing or massively parallel sequencing is any of several high-throughput approaches to DNA sequencing using the concept of massively parallel processing; it is also called next-generation sequencing (NGS) or second-generation sequencing. Some of these technologies emerged between 1993 and 1998 and have been commercially available since 2005. These technologies use miniaturized and parallelized platforms for sequencing of 1 million to 43 billion short reads per instrument run.
Cell-free fetal DNA (cffDNA) is fetal DNA that circulates freely in the maternal blood. Maternal blood is sampled by venipuncture. Analysis of cffDNA is a method of non-invasive prenatal diagnosis frequently ordered for pregnant women of advanced maternal age. Two hours after delivery, cffDNA is no longer detectable in maternal blood.

In the field of cellular biology, single-cell analysis and subcellular analysis is the study of genomics, transcriptomics, proteomics, metabolomics and cell–cell interactions at the single cell level. The concept of single-cell analysis originated in the 1970s. Before the discovery of heterogeneity, single-cell analysis mainly referred to the analysis or manipulation of an individual cell in a bulk population of cells at a particular condition using optical or electronic microscope. To date, due to the heterogeneity seen in both eukaryotic and prokaryotic cell populations, analyzing a single cell makes it possible to discover mechanisms not seen when studying a bulk population of cells. Technologies such as fluorescence-activated cell sorting (FACS) allow the precise isolation of selected single cells from complex samples, while high throughput single cell partitioning technologies, enable the simultaneous molecular analysis of hundreds or thousands of single unsorted cells; this is particularly useful for the analysis of transcriptome variation in genotypically identical cells, allowing the definition of otherwise undetectable cell subtypes. The development of new technologies is increasing our ability to analyze the genome and transcriptome of single cells, as well as to quantify their proteome and metabolome. Mass spectrometry techniques have become important analytical tools for proteomic and metabolomic analysis of single cells. Recent advances have enabled quantifying thousands of protein across hundreds of single cells, and thus make possible new types of analysis. In situ sequencing and fluorescence in situ hybridization (FISH) do not require that cells be isolated and are increasingly being used for analysis of tissues.

Circulating tumor DNA (ctDNA) is tumor-derived fragmented DNA in the bloodstream that is not associated with cells. ctDNA should not be confused with cell-free DNA (cfDNA), a broader term which describes DNA that is freely circulating in the bloodstream, but is not necessarily of tumor origin. Because ctDNA may reflect the entire tumor genome, it has gained traction for its potential clinical utility; "liquid biopsies" in the form of blood draws may be taken at various time points to monitor tumor progression throughout the treatment regimen.
CAncer Personalized Profiling by deep Sequencing (CAPP-Seq) is a next-generation sequencing based method used to quantify circulating DNA in cancer (ctDNA). The method was introduced in 2014 by Ash Alizadeh and Maximilian Diehn’s laboratories at Stanford, as a tool for measuring Cell-free tumor DNA which is released from dead tumor cells into the blood and thus may reflect the entire tumor genome. This method can be generalized for any cancer type that is known to have recurrent mutations. CAPP-Seq can detect one molecule of mutant DNA in 10,000 molecules of healthy DNA. The original method was further refined in 2016 for ultra sensitive detection through integration of multiple error suppression strategies, termed integrated Digital Error Suppression (iDES). The use of ctDNA in this technique should not be confused with circulating tumor cells (CTCs); these are two different entities.
Droplet-based microfluidics manipulate discrete volumes of fluids in immiscible phases with low Reynolds number and laminar flow regimes. Interest in droplet-based microfluidics systems has been growing substantially in past decades. Microdroplets offer the feasibility of handling miniature volumes of fluids conveniently, provide better mixing, encapsulation, sorting, sensing and are suitable for high throughput experiments. Two immiscible phases used for the droplet based systems are referred to as the continuous phase and dispersed phase.
Luis Alberto Diaz, Jr. is the Head of the Division of Solid Tumor Oncology in Memorial Sloan Kettering’s Department of Medicine.
References
- ↑ Diehl, Frank; Smergeliene, Edita (September 1, 2013). "BEAMing for Cancer: Detecting Tumor Mutations in Peripheral Blood Using Digital PCR".
- 1 2 3 Diehl, Frank; Li, Meng; Dressman, Devin; He, Yiping; Shen, Dong; Szabo, Steve; Diaz, Luis A.; Goodman, Steven N.; David, Kerstin A. (2005-11-08). "Detection and quantification of mutations in the plasma of patients with colorectal tumors". Proceedings of the National Academy of Sciences of the United States of America. 102 (45): 16368–16373. Bibcode:2005PNAS..10216368D. doi: 10.1073/pnas.0507904102 . ISSN 0027-8424. PMC 1283450 . PMID 16258065.
- ↑ Li, Meng; Diehl, Frank; Dressman, Devin; Vogelstein, Bert; Kinzler, Kenneth W (2006). "BEAMing up for detection and quantification of rare sequence variants". Nature Methods. 3 (2): 95–97. doi:10.1038/nmeth850. PMID 16432518. S2CID 13590593.
- ↑ Wan, Jonathan C. M.; Massie, Charles; Garcia-Corbacho, Javier; Mouliere, Florent; Brenton, James D.; Caldas, Carlos; Pacey, Simon; Baird, Richard; Rosenfeld, Nitzan (2017). "Liquid biopsies come of age: towards implementation of circulating tumour DNA". Nature Reviews Cancer. 17 (4): 223–238. doi:10.1038/nrc.2017.7. PMID 28233803. S2CID 4561229.
- 1 2 Morley, Alexander A. (2014). "Digital PCR: A brief history". Biomolecular Detection and Quantification. 1 (1): 1–2. doi:10.1016/j.bdq.2014.06.001. PMC 5129430 . PMID 27920991.
- 1 2 Vogelstein, Bert; Kinzler, Kenneth W. (1999-08-03). "Digital PCR". Proceedings of the National Academy of Sciences of the United States of America. 96 (16): 9236–9241. Bibcode:1999PNAS...96.9236V. doi: 10.1073/pnas.96.16.9236 . ISSN 0027-8424. PMC 17763 . PMID 10430926.
- ↑ Dressman, Devin; Yan, Hai; Traverso, Giovanni; Kinzler, Kenneth W.; Vogelstein, Bert (2003-07-22). "Transforming single DNA molecules into fluorescent magnetic particles for detection and enumeration of genetic variations". Proceedings of the National Academy of Sciences. 100 (15): 8817–8822. Bibcode:2003PNAS..100.8817D. doi: 10.1073/pnas.1133470100 . ISSN 0027-8424. PMC 166396 . PMID 12857956.
- ↑ Diehl, Frank; Schmidt, Kerstin; Choti, Michael A; Romans, Katharine; Goodman, Steven; Li, Meng; Thornton, Katherine; Agrawal, Nishant; Sokoll, Lori (2008). "Circulating mutant DNA to assess tumor dynamics". Nature Medicine. 14 (9): 985–990. doi:10.1038/nm.1789. PMC 2820391 . PMID 18670422.
- ↑ "UPDATE: Japan's Sysmex Acquires Germany's Inostics, Partec". GenomeWeb. Retrieved 2017-10-17.