Related Research Articles

Rhodopsin, also known as visual purple, is a protein encoded by the RHO gene and a G-protein-coupled receptor (GPCR). It is a light-sensitive receptor protein that triggers visual phototransduction in rods. Rhodopsin mediates dim light vision and thus is extremely sensitive to light. When rhodopsin is exposed to light, it immediately photobleaches. In humans, it is regenerated fully in about 30 minutes, after which the rods are more sensitive. Defects in the rhodopsin gene cause eye diseases such as retinitis pigmentosa and congenital stationary night blindness.

Retinitis pigmentosa (RP) is a member of a group of genetic disorders called inherited retinal dystrophy (IRD) that cause loss of vision. Symptoms include trouble seeing at night and decreasing peripheral vision. As peripheral vision worsens, people may experience "tunnel vision". Complete blindness is uncommon. Onset of symptoms is generally gradual and often begins in childhood.

Usher syndrome, also known as Hallgren syndrome, Usher–Hallgren syndrome, retinitis pigmentosa–dysacusis syndrome or dystrophia retinae dysacusis syndrome, is a rare genetic disorder caused by a mutation in any one of at least 11 genes resulting in a combination of hearing loss and visual impairment. It is the most common cause of deafblindness and is at present incurable.

Choroideremia is a rare, X-linked recessive form of hereditary retinal degeneration that affects roughly 1 in 50,000 males. The disease causes a gradual loss of vision, starting with childhood night blindness, followed by peripheral vision loss and progressing to loss of central vision later in life. Progression continues throughout the individual's life, but both the rate of change and the degree of visual loss are variable among those affected, even within the same family.

The photoreceptor cell-specific nuclear receptor (PNR), also known as NR2E3, is a protein that in humans is encoded by the NR2E3 gene. PNR is a member of the nuclear receptor super family of intracellular transcription factors.

X-linked retinitis pigmentosa GTPase regulator is a GTPase-binding protein that in humans is encoded by the RPGR gene. The gene is located on the X-chromosome and is commonly associated with X-linked retinitis pigmentosa (XLRP). In photoreceptor cells, RPGR is localized in the connecting cilium which connects the protein-synthesizing inner segment to the photosensitive outer segment and is involved in the modulation of cargo trafficked between the two segments.

PRP31 pre-mRNA processing factor 31 homolog , also known as PRPF31, is a protein which in humans is encoded by the PRPF31 gene.

Retinaldehyde-binding protein 1 (RLBP1) also known as cellular retinaldehyde-binding protein (CRALBP) is a 36-kD water-soluble protein that in humans is encoded by the RLBP1 gene.

Protein XRP2 is a protein that in humans is encoded by the RP2 gene.
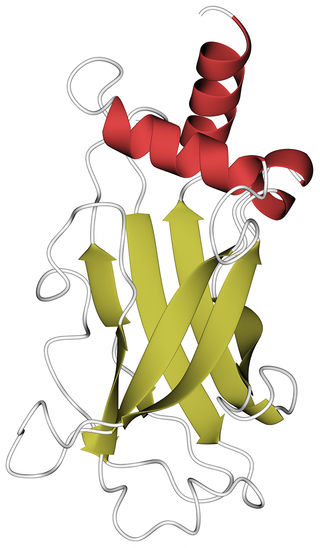
X-linked retinitis pigmentosa GTPase regulator-interacting protein 1 is a protein in the ciliary transition zone that in humans is encoded by the RPGRIP1 gene. RPGRIP1 is a multi-domain protein containing a coiled-coil domain at the N-terminus, two C2 domains and a C-terminal RPGR-interacting domain (RID). Defects in the gene result in the Leber congenital amaurosis (LCA) syndrome and in the eye disease glaucoma.
Thaddeus P. Dryja is an American ophthalmologist and geneticist known for his role in the 1986 discovery of the retinoblastoma (Rb) tumor suppressor gene. and the 1990 discovery of mutations in the rhodopsin gene as the cause of autosomal dominant retinitis pigmentosa . He was the David G. Cogan Professor of Ophthalmology at Harvard University and was the Global Head of Ophthalmology Research at Novartis. He was elected a member of the National Academy of Sciences in 1996.

The Llura Liggett Gund Award honors researchers for career achievements that have significantly advanced the research and development of preventions, treatments and cures for eye disease.
Retinal gene therapy holds a promise in treating different forms of non-inherited and inherited blindness.
Stephen H. Tsang is an American ophthalmologist and geneticist. He is currently a Professor of Ophthalmology, and Pathology and Cell Biology at Columbia University Irving Medical Center in New York.
The Applied Genetic Technologies Corporation is a publicly traded biotechnology company that is part of the NASDAQ Biotechnology Index. It was founded in 1999 and has its headquarters in Alachua, Florida. In late June 2019, the company announced the appointment of Global Clinical and Medical Affairs Veteran, Theresa G.H. Heah, M.D., M.B.A., to Join as Chief Medical Officer.

Robert E. MacLaren is a British ophthalmologist who has led pioneering work in the treatment of blindness caused by diseases of the retina. He is Professor of Ophthalmology at the University of Oxford and Honorary Professor of Ophthalmology at the UCL Institute of Ophthalmology. He is a Consultant Ophthalmologist at the Oxford Eye Hospital. He is also an Honorary Consultant Vitreo-retinal Surgeon at the Moorfields Eye Hospital. MacLaren is an NIHR Senior Investigator, or lead researcher, for the speciality of Ophthalmology. In addition, he is a member of the research committee of Euretina: the European Society of Retina specialists, Fellow of Merton College, in Oxford and a Fellow of the Higher Education Academy.
Occult macular dystrophy (OMD) is a rare inherited degradation of the retina, characterized by progressive loss of function in the most sensitive part of the central retina (macula), the location of the highest concentration of light-sensitive cells (photoreceptors) but presenting no visible abnormality. "Occult" refers to the degradation in the fundus being difficult to discern. The disorder is called "dystrophy" instead of "degradation" to distinguish its genetic origin from other causes, such as age. OMD was first reported by Y. Miyake et al. in 1989.

Paul A. Sieving is a former director of the National Eye Institute, part of the U.S. National Institutes of Health. Prior to joining the NIH in 2001, he served on the faculty of the University of Michigan Medical School as the Paul R. Lichter Professor of Ophthalmic Genetics. He also was the founding director of the Center for Retinal and Macular Degeneration in the university's Department of Ophthalmology and Visual Sciences.
William Anthony Beltran is a French–American ophthalmologist. He is a professor of ophthalmology in the Department of Clinical Sciences and Advanced Medicine and director of the Division of Experimental Retinal Therapies at the University of Pennsylvania School of Veterinary Medicine. In 2020, Beltran was elected a Member of the National Academy of Medicine for his research focus on inherited retinal degeneration.
Intravitreal gene therapy represents an approach to treating retinal diseases by delivering therapeutic genes directly into the vitreous humor of the eye. This method uses a viral vector, often an adeno-associated virus (AAV), to carry genetic material into retinal cells. Once inside, the therapeutic genes are expressed to address genetic deficiencies or modify biological pathways, offering a long-term or potentially permanent treatment for conditions like wet age-related macular degeneration (AMD), diabetic macular edema, and inherited retinal dystrophies. Unlike traditional therapies requiring frequent injections, intravitreal gene therapy aims to reduce the treatment burden while improving efficacy potentially providing lifelong benefit.
References
- ↑ Michaelides, Michel; Xu, Jialin; Wang, Dai; Wong, Peggy; Fung, Albert; Forbes, Alexandra; Naylor, Stuart; Zeldin, Robert; Parker, Maria A; Weleber, Richard; Guimaraes, Thales Antonio Cabral de; Besirli, Cagri; Yang, Yesa; Bainbridge, James (2022-06-01). "AAV5-RPGR (botaretigene sparoparvovec) gene therapy for X-linked retinitis pigmentosa (XLRP) demonstrates localized improvements in static perimetry". Investigative Ophthalmology & Visual Science. 63 (7). The Association for Research in Vision and Ophthalmology: 3846. ISSN 1552-5783 . Retrieved 2023-12-05.
- ↑ "Humoral immune response to AAV5-RPGR (botaretigene sparoparvovec) gene therapy in RPGR-associated X-linked retinitis pigmentosa". Investigative Ophthalmology & Visual Science.