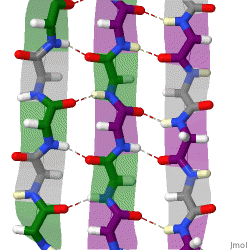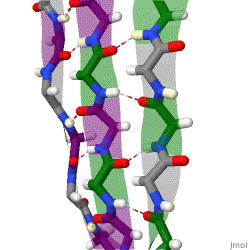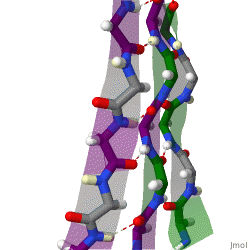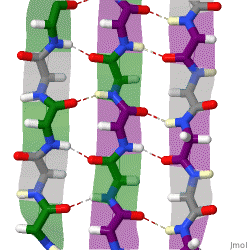
The beta sheet is a common motif of the regular protein secondary structure. Beta sheets consist of beta strands (β-strands) connected laterally by at least two or three backbone hydrogen bonds, forming a generally twisted, pleated sheet. A β-strand is a stretch of polypeptide chain typically 3 to 10 amino acids long with backbone in an extended conformation. The supramolecular association of β-sheets has been implicated in the formation of the fibrils and protein aggregates observed in amyloidosis, Alzheimer's disease and other proteinopathies.

Protein secondary structure is the local spatial conformation of the polypeptide backbone excluding the side chains. The two most common secondary structural elements are alpha helices and beta sheets, though beta turns and omega loops occur as well. Secondary structure elements typically spontaneously form as an intermediate before the protein folds into its three dimensional tertiary structure.

In molecular biology, post-translational modification (PTM) is the covalent process of changing proteins following protein biosynthesis. PTMs may involve enzymes or occur spontaneously. Proteins are created by ribosomes, which translate mRNA into polypeptide chains, which may then change to form the mature protein product. PTMs are important components in cell signalling, as for example when prohormones are converted to hormones.

Protein structure prediction is the inference of the three-dimensional structure of a protein from its amino acid sequence—that is, the prediction of its secondary and tertiary structure from primary structure. Structure prediction is different from the inverse problem of protein design. Protein structure prediction is one of the most important goals pursued by computational biology; it is important in medicine and biotechnology.
In a chain-like biological molecule, such as a protein or nucleic acid, a structural motif is a common three-dimensional structure that appears in a variety of different, evolutionarily unrelated molecules. A structural motif does not have to be associated with a sequence motif; it can be represented by different and completely unrelated sequences in different proteins or RNA.

Picornaviruses are a group of related nonenveloped RNA viruses which infect vertebrates including fish, mammals, and birds. They are viruses that represent a large family of small, positive-sense, single-stranded RNA viruses with a 30 nm icosahedral capsid. The viruses in this family can cause a range of diseases including the common cold, poliomyelitis, meningitis, hepatitis, and paralysis.

Structural alignment attempts to establish homology between two or more polymer structures based on their shape and three-dimensional conformation. This process is usually applied to protein tertiary structures but can also be used for large RNA molecules. In contrast to simple structural superposition, where at least some equivalent residues of the two structures are known, structural alignment requires no a priori knowledge of equivalent positions. Structural alignment is a valuable tool for the comparison of proteins with low sequence similarity, where evolutionary relationships between proteins cannot be easily detected by standard sequence alignment techniques. Structural alignment can therefore be used to imply evolutionary relationships between proteins that share very little common sequence. However, caution should be used in using the results as evidence for shared evolutionary ancestry because of the possible confounding effects of convergent evolution by which multiple unrelated amino acid sequences converge on a common tertiary structure.

Protein structure is the three-dimensional arrangement of atoms in an amino acid-chain molecule. Proteins are polymers – specifically polypeptides – formed from sequences of amino acids, which are the monomers of the polymer. A single amino acid monomer may also be called a residue, which indicates a repeating unit of a polymer. Proteins form by amino acids undergoing condensation reactions, in which the amino acids lose one water molecule per reaction in order to attach to one another with a peptide bond. By convention, a chain under 30 amino acids is often identified as a peptide, rather than a protein. To be able to perform their biological function, proteins fold into one or more specific spatial conformations driven by a number of non-covalent interactions, such as hydrogen bonding, ionic interactions, Van der Waals forces, and hydrophobic packing. To understand the functions of proteins at a molecular level, it is often necessary to determine their three-dimensional structure. This is the topic of the scientific field of structural biology, which employs techniques such as X-ray crystallography, NMR spectroscopy, cryo-electron microscopy (cryo-EM) and dual polarisation interferometry, to determine the structure of proteins.
The terms glycans and polysaccharides are defined by IUPAC as synonyms meaning "compounds consisting of a large number of monosaccharides linked glycosidically". However, in practice the term glycan may also be used to refer to the carbohydrate portion of a glycoconjugate, such as a glycoprotein, glycolipid, or a proteoglycan, even if the carbohydrate is only an oligosaccharide. Glycans usually consist solely of O-glycosidic linkages of monosaccharides. For example, cellulose is a glycan composed of β-1,4-linked D-glucose, and chitin is a glycan composed of β-1,4-linked N-acetyl-D-glucosamine. Glycans can be homo- or heteropolymers of monosaccharide residues, and can be linear or branched.
A DNA-binding domain (DBD) is an independently folded protein domain that contains at least one structural motif that recognizes double- or single-stranded DNA. A DBD can recognize a specific DNA sequence or have a general affinity to DNA. Some DNA-binding domains may also include nucleic acids in their folded structure.

Transcriptional modification or co-transcriptional modification is a set of biological processes common to most eukaryotic cells by which an RNA primary transcript is chemically altered following transcription from a gene to produce a mature, functional RNA molecule that can then leave the nucleus and perform any of a variety of different functions in the cell. There are many types of post-transcriptional modifications achieved through a diverse class of molecular mechanisms.

Histone H2A is one of the five main histone proteins involved in the structure of chromatin in eukaryotic cells.

In protein structures, a beta barrel(β barrel) is a beta sheet composed of tandem repeats that twists and coils to form a closed toroidal structure in which the first strand is bonded to the last strand. Beta-strands in many beta-barrels are arranged in an antiparallel fashion. Beta barrel structures are named for resemblance to the barrels used to contain liquids. Most of them are water-soluble outer membrane proteins and frequently bind hydrophobic ligands in the barrel center, as in lipocalins. Others span cell membranes and are commonly found in porins. Porin-like barrel structures are encoded by as many as 2–3% of the genes in Gram-negative bacteria. It has been shown that more than 600 proteins with various function such as oxidase, dismutase, and amylase contain the beta barrel structure.
A turn is an element of secondary structure in proteins where the polypeptide chain reverses its overall direction.

The TIM barrel, also known as an alpha/beta barrel, is a conserved protein fold consisting of eight alpha helices (α-helices) and eight parallel beta strands (β-strands) that alternate along the peptide backbone. The structure is named after triose-phosphate isomerase, a conserved metabolic enzyme. TIM barrels are ubiquitous, with approximately 10% of all enzymes adopting this fold. Further, five of seven enzyme commission (EC) enzyme classes include TIM barrel proteins. The TIM barrel fold is evolutionarily ancient, with many of its members possessing little similarity today, instead falling within the twilight zone of sequence similarity.

The EF hand is a helix–loop–helix structural domain or motif found in a large family of calcium-binding proteins.
The omega loop is a non-regular protein structural motif, consisting of a loop of six or more amino acid residues and any amino acid sequence. The defining characteristic is that residues that make up the beginning and end of the loop are close together in space with no intervening lengths of regular secondary structural motifs. It is named after its shape, which resembles the upper-case Greek letter Omega (Ω).
Loop modeling is a problem in protein structure prediction requiring the prediction of the conformations of loop regions in proteins with or without the use of a structural template. Computer programs that solve these problems have been used to research a broad range of scientific topics from ADP to breast cancer. Because protein function is determined by its shape and the physiochemical properties of its exposed surface, it is important to create an accurate model for protein/ligand interaction studies. The problem arises often in homology modeling, where the tertiary structure of an amino acid sequence is predicted based on a sequence alignment to a template, or a second sequence whose structure is known. Because loops have highly variable sequences even within a given structural motif or protein fold, they often correspond to unaligned regions in sequence alignments; they also tend to be located at the solvent-exposed surface of globular proteins and thus are more conformationally flexible. Consequently, they often cannot be modeled using standard homology modeling techniques. More constrained versions of loop modeling are also used in the data fitting stages of solving a protein structure by X-ray crystallography, because loops can correspond to regions of low electron density and are therefore difficult to resolve.

In molecular biology, a protein domain is a region of a protein's polypeptide chain that is self-stabilizing and that folds independently from the rest. Each domain forms a compact folded three-dimensional structure. Many proteins consist of several domains, and a domain may appear in a variety of different proteins. Molecular evolution uses domains as building blocks and these may be recombined in different arrangements to create proteins with different functions. In general, domains vary in length from between about 50 amino acids up to 250 amino acids in length. The shortest domains, such as zinc fingers, are stabilized by metal ions or disulfide bridges. Domains often form functional units, such as the calcium-binding EF hand domain of calmodulin. Because they are independently stable, domains can be "swapped" by genetic engineering between one protein and another to make chimeric proteins.

General bacterial porins are a family of porin proteins from the outer membranes of Gram-negative bacteria. The porins act as molecular filters for hydrophilic compounds. They are responsible for the 'molecular sieve' properties of the outer membrane. Porins form large water-filled channels which allow the diffusion of hydrophilic molecules into the periplasmic space. Some porins form general diffusion channels that allow any solute up to a certain size to cross the membrane, while other porins are specific for one particular solute and contain a binding site for that solute inside the pores. As porins are the major outer membrane proteins, they also serve as receptor sites for the binding of phages and bacteriocins.