A representation of the 3D structure of the protein myoglobin showing turquoise α-helices. This protein was the first to have its structure solved by X-ray crystallography. Toward the right-center among the coils, a prosthetic group called a heme group (shown in gray) with a bound oxygen molecule (red).
A linear chain of amino acid residues is called a polypeptide. A protein contains at least one long polypeptide. Short polypeptides, containing less than 20–30 residues, are rarely considered to be proteins and are commonly called peptides. The individual amino acid residues are bonded together by peptide bonds and adjacent amino acid residues. The sequence of amino acid residues in a protein is defined by the sequence of a gene, which is encoded in the genetic code. In general, the genetic code specifies 20 standard amino acids; but in certain organisms the genetic code can include selenocysteine and—in certain archaea—pyrrolysine. Shortly after or even during synthesis, the residues in a protein are often chemically modified by post-translational modification, which alters the physical and chemical properties, folding, stability, activity, and ultimately, the function of the proteins. Some proteins have non-peptide groups attached, which can be called prosthetic groups or cofactors. Proteins can work together to achieve a particular function, and they often associate to form stable protein complexes.
Once formed, proteins only exist for a certain period and are then degraded and recycled by the cell's machinery through the process of protein turnover. A protein's lifespan is measured in terms of its half-life and covers a wide range. They can exist for minutes or years with an average lifespan of 1–2 days in mammalian cells. Abnormal or misfolded proteins are degraded more rapidly either due to being targeted for destruction or due to being unstable.
Like other biological macromolecules such as polysaccharides and nucleic acids, proteins are essential parts of organisms and participate in virtually every process within cells. Many proteins are enzymes that catalyse biochemical reactions and are vital to metabolism. Some proteins have structural or mechanical functions, such as actin and myosin in muscle, and the cytoskeleton's scaffolding proteins that maintain cell shape. Other proteins are important in cell signaling, immune responses, cell adhesion, and the cell cycle. In animals, proteins are needed in the diet to provide the essential amino acids that cannot be synthesized. Digestion breaks the proteins down for metabolic use.
Proteins have been studied and recognized since the 1700s by Antoine Fourcroy and others,[1][2] who often collectively called them "albumins", or "albuminous materials" (Eiweisskörper, in German).[2]Gluten, for example, was first separated from wheat in published research around 1747, and later determined to exist in many plants.[1] In 1789, Antoine Fourcroy recognized three distinct varieties of animal proteins: albumin, fibrin, and gelatin.[3] Vegetable (plant) proteins studied in the late 1700s and early 1800s included gluten, plant albumin, gliadin, and legumin.[1]
Proteins were first described by the Dutch chemist Gerardus Johannes Mulder and named by the Swedish chemist Jöns Jacob Berzelius in 1838.[4][5] Mulder carried out elemental analysis of common proteins and found that nearly all proteins had the same empirical formula, C400H620N100O120P1S1.[6] He came to the erroneous conclusion that they might be composed of a single type of (very large) molecule. The term "protein" to describe these molecules was proposed by Mulder's associate Berzelius; protein is derived from the Greek word πρώτειος (proteios), meaning "primary",[7] "in the lead", or "standing in front",[2] + -in. Mulder went on to identify the products of protein degradation such as the amino acidleucine for which he found a (nearly correct) molecular weight of 131Da.[6]
The difficulty in purifying proteins impeded work by early protein biochemists. Proteins could be obtained in large quantities from blood, egg whites, and keratin, but individual proteins were unavailable. In the 1950s, the Armour Hot Dog Company purified 1kg of bovine pancreatic ribonuclease A and made it freely available to scientists. This gesture helped ribonuclease A become a major target for biochemical study for the following decades.[6]
Polypeptides
polypeptide
The understanding of proteins as polypeptides, or chains of amino acids, came through the work of Franz Hofmeister and Hermann Emil Fischer in 1902.[13][14] The central role of proteins as enzymes in living organisms that catalyzed reactions was not fully appreciated until 1926, when James B. Sumner showed that the enzyme urease was in fact a protein.[15]
The first protein to have its amino acid chain sequenced was insulin, by Frederick Sanger, in 1949. Sanger correctly determined the amino acid sequence of insulin, thus conclusively demonstrating that proteins consisted of linear polymers of amino acids rather than branched chains, colloids, or cyclols.[21] He won the Nobel Prize for this achievement in 1958.[22]Christian Anfinsen's studies of the oxidative folding process of ribonuclease A, for which he won the nobel prize in 1972, solidified the thermodynamic hypothesis of protein folding, according to which the folded form of a protein represents its free energy minimum.[23][24]
Proteins are primarily classified by sequence and structure, although other classifications are commonly used. Especially for enzymes the EC number system provides a functional classification scheme.[31] Similarly, gene ontology classifies both genes and proteins by their biological and biochemical function, and by their intracellular location.[32]
Sequence similarity is used to classify proteins both in terms of evolutionary and functional similarity. This may use either whole proteins or protein domains, especially in multi-domain proteins. Protein domains allow protein classification by a combination of sequence, structure and function, and they can be combined in many ways. In an early study of 170,000 proteins, about two-thirds were assigned at least one domain, with larger proteins containing more domains (e.g. proteins larger than 600 amino acids having an average of more than 5 domains).[33]
Biochemistry
Chemical structure of the peptide bond (bottom) and the three-dimensional structure of a peptide bond between an alanine and an adjacent amino acid (top/inset). The bond itself is made of the CHON elements.Resonance structures of the peptide bond that links individual amino acids to form a protein polymer
Most proteins consist of linear polymers built from series of up to 20 L-α-amino acids. All proteinogenic amino acids have a common structure where an α-carbon is bonded to an amino group, a carboxyl group, and a variable side chain. Only proline differs from this basic structure as its side chain is cyclical, bonding to the amino group, limiting protein chain flexibility.[34] The side chains of the standard amino acids have a variety of chemical structures and properties, and it is the combined effect of all amino acids that determines its three-dimensional structure and chemical reactivity.[35]
The amino acids in a polypeptide chain are linked by peptide bonds between amino and carboxyl group. An individual amino acid in a chain is called a residue, and the linked series of carbon, nitrogen, and oxygen atoms are known as the main chain or protein backbone.[36]:19 The peptide bond has two resonance forms that confer some double-bond character to the backbone. The alpha carbons are roughly coplanar with the nitrogen and the carbonyl (C=O) group. The other two dihedral angles in the peptide bond determine the local shape assumed by the protein backbone. One consequence of the N-C(O) double bond character is that proteins are somewhat rigid.[36]:31 A polypeptide chain ends with a free amino group, known as the N-terminus or amino terminus, and a free carboxyl group, known as the C-terminus or carboxy terminus.[37] By convention, peptide sequences are written N-terminus to C-terminus, correlating with the order in which proteins are synthesized by ribosomes.[37][38]
The words protein, polypeptide, and peptide are a little ambiguous and can overlap in meaning. Protein is generally used to refer to the complete biological molecule in a stable conformation, whereas peptide is generally reserved for a short amino acid oligomers often lacking a stable 3D structure. But the boundary between the two is not well defined and usually lies near 20–30 residues.[39]
A typical bacterial cell, e.g. E. coli and Staphylococcus aureus, is estimated to contain about 2 million proteins. Smaller bacteria, such as Mycoplasma or spirochetes contain fewer molecules, on the order of 50,000 to 1 million. By contrast, eukaryotic cells are larger and thus contain much more protein. For instance, yeast cells have been estimated to contain about 50 million proteins and human cells on the order of 1 to 3 billion.[43] The concentration of individual protein copies ranges from a few molecules per cell up to 20 million.[44] Not all genes coding proteins are expressed in most cells and their number depends on, for example, cell type and external stimuli. For instance, of the 20,000 or so proteins encoded by the human genome, only 6,000 are detected in lymphoblastoid cells.[45] The most abundant protein in nature is thought to be RuBisCO, an enzyme that catalyzes the incorporation of carbon dioxide into organic matter in photosynthesis. Plants can consist of as much as 1% by weight of this enzyme.[46]
Synthesis
Biosynthesis
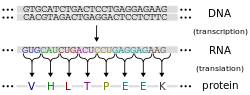
A ribosome produces a protein using mRNA as templateThe DNA sequence of a gene encodes the amino acid sequence of a protein
Proteins are assembled from amino acids using information encoded in genes. Each protein has its own unique amino acid sequence that is specified by the nucleotide sequence of the gene encoding this protein. The genetic code is a set of three-nucleotide sets called codons and each three-nucleotide combination designates an amino acid, for example AUG (adenine–uracil–guanine) is the code for methionine. Because DNA contains four nucleotides, the total number of possible codons is 64; hence, there is some redundancy in the genetic code, with some amino acids specified by more than one codon.[42]:1002–42 Genes encoded in DNA are first transcribed into pre-messenger RNA (mRNA) by proteins such as RNA polymerase. Most organisms then process the pre-mRNA (a primary transcript) using various forms of post-transcriptional modification to form the mature mRNA, which is then used as a template for protein synthesis by the ribosome. In prokaryotes the mRNA may either be used as soon as it is produced, or be bound by a ribosome after having moved away from the nucleoid. In contrast, eukaryotes make mRNA in the cell nucleus and then translocate it across the nuclear membrane into the cytoplasm, where protein synthesis then takes place. The rate of protein synthesis is higher in prokaryotes than eukaryotes and can reach up to 20 amino acids per second.[47]
The process of synthesizing a protein from an mRNA template is known as translation. The mRNA is loaded onto the ribosome and is read three nucleotides at a time by matching each codon to its base pairinganticodon located on a transfer RNA molecule, which carries the amino acid corresponding to the codon it recognizes. The enzyme aminoacyl tRNA synthetase "charges" the tRNA molecules with the correct amino acids. The growing polypeptide is often termed the nascent chain. Proteins are always biosynthesized from N-terminus to C-terminus.[42]:1002–42
The size of a synthesized protein can be measured by the number of amino acids it contains and by its total molecular mass, which is normally reported with the unit dalton (Da), or its derivative unit kilodalton (kDa). The average size of a protein increases from Archaea to Bacteria to Eukaryote (283, 311, 438 residues and 31, 34, 49kDa respectively) due to a bigger number of protein domains constituting proteins in higher organisms.[48] For instance, yeast proteins are on average 466 amino acids long and 53 kDa in mass.[39] The largest known proteins are the titins, a component of the musclesarcomere, with a molecular mass of almost 3000kDa and a total length of almost 27000 amino acids.[49]
Short proteins can be synthesized chemically by a family of peptide synthesis methods. These rely on organic synthesis techniques such as chemical ligation to produce peptides in high yield.[50] Chemical synthesis allows for the introduction of non-natural amino acids into polypeptide chains, such as attachment of fluorescent probes to amino acid side chains.[51] These methods are useful in laboratory biochemistry and cell biology, though generally not for commercial applications. Chemical synthesis is inefficient for polypeptides longer than about 300 amino acids, and the synthesized proteins may not readily assume their native tertiary structure. Most chemical synthesis methods proceed from C-terminus to N-terminus, opposite the biological reaction.[52]
Structure
The crystal structure of the chaperonin, a huge protein complex. A single protein subunit is highlighted. Chaperonins assist protein folding.Three possible representations of the three-dimensional structure of the protein triose phosphate isomerase. Left: All-atom representation colored by atom type. Middle: Simplified representation illustrating the backbone conformation, colored by secondary structure. Right: Solvent-accessible surface representation colored by residue type (acidic residues red, basic residues blue, polar residues green, nonpolar residues white).
Most proteins fold into unique 3D structures. The shape into which a protein naturally folds is known as its native conformation.[36]:36 Although many proteins can fold unassisted, simply through the chemical properties of their amino acids, others require the aid of molecular chaperones to fold into their native states.[36]:37 Biochemists often refer to four distinct aspects of a protein's structure:[36]:30–34
Secondary structure: regularly repeating local structures stabilized by hydrogen bonds. The most common examples are the α-helix, β-sheet and turns. Because secondary structures are local, many regions of distinct secondary structure can be present in the same protein molecule.
Tertiary structure: the overall shape of a single protein molecule; the spatial relationship of the secondary structures to one another. Tertiary structure is generally stabilized by nonlocal interactions, most commonly the formation of a hydrophobic core, but also through salt bridges, hydrogen bonds, disulfide bonds, and even post-translational modifications. The term "tertiary structure" is often used as synonymous with the term fold. The tertiary structure is what controls the basic function of the protein.
Quinary structure: the signatures of protein surface that organize the crowded cellular interior. Quinary structure is dependent on transient, yet essential, macromolecular interactions that occur inside living cells.
Proteins are not entirely rigid molecules. In addition to these levels of structure, proteins may shift between several related structures while they perform their functions. In the context of these functional rearrangements, these tertiary or quaternary structures are usually referred to as "conformations", and transitions between them are called conformational changes. Such changes are often induced by the binding of a substrate molecule to an enzyme's active site, or the physical region of the protein that participates in chemical catalysis. In solution, protein structures vary because of thermal vibration and collisions with other molecules.[42]:368–75
Proteins can be informally divided into three main classes, which correlate with typical tertiary structures: globular proteins, fibrous proteins, and membrane proteins. Almost all globular proteins are soluble and many are enzymes. Fibrous proteins are often structural, such as collagen, the major component of connective tissue, or keratin, the protein component of hair and nails. Membrane proteins often serve as receptors or provide channels for polar or charged molecules to pass through the cell membrane.[42]:165–85
A special case of intramolecular hydrogen bonds within proteins, poorly shielded from water attack and hence promoting their own dehydration, are called dehydrons.[53]
Many proteins are composed of several protein domains, i.e. segments of a protein that fold into distinct structural units.[54]:134 Domains usually have specific functions, such as enzymatic activities (e.g. kinase) or they serve as binding modules.[54]:155–156
Protein domains vs. motifs. Protein domains (such as the EVH1 domain) are functional units within proteins that fold into defined 3D structures. Motifs are usually short sequences with specific functions but without a stable 3D structure. Many motifs are binding sites for other proteins (such as the red and green bars shown here in the context of a VASP protein).
Sequence motif
Short amino acid sequences within proteins often act as recognition sites for other proteins.[56] For instance, SH3 domains typically bind to short PxxP motifs (i.e. 2 prolines [P], separated by two unspecified amino acids [x], although the surrounding amino acids may determine the exact binding specificity). Many such motifs has been collected in the Eukaryotic Linear Motif (ELM) database.[57]
Cellular functions
Proteins are the chief actors within the cell, said to be carrying out the duties specified by the information encoded in genes.[39] With the exception of certain types of RNA, most other biological molecules are relatively inert elements upon which proteins act. Proteins make up half the dry weight of an Escherichia coli cell, whereas other macromolecules such as DNA and RNA make up only 3% and 20%, respectively.[58] The set of proteins expressed in a particular cell or cell type is known as its proteome.[54]:120
The enzyme hexokinase is shown as a conventional ball-and-stick molecular model. To scale in the top right-hand corner are two of its substrates, ATP and glucose.
The chief characteristic of proteins that allows their diverse set of functions is their ability to bind other molecules specifically and tightly. The region of the protein responsible for binding another molecule is known as the binding site and is often a depression or "pocket" on the molecular surface. This binding ability is mediated by the tertiary structure of the protein, which defines the binding site pocket, and by the chemical properties of the surrounding amino acids' side chains. Protein binding can be extraordinarily tight and specific; for example, the ribonuclease inhibitor protein binds to human angiogenin with a sub-femtomolar dissociation constant (<10−15M) but does not bind at all to its amphibian homolog onconase (>1M). Extremely minor chemical changes such as the addition of a single methyl group to a binding partner can sometimes suffice to nearly eliminate binding; for example, the aminoacyl tRNA synthetase specific to the amino acid valine discriminates against the very similar side chain of the amino acid isoleucine.[59]
Proteins can bind to other proteins as well as to small-molecule substrates. When proteins bind specifically to other copies of the same molecule, they can oligomerize to form fibrils; this process occurs often in structural proteins that consist of globular monomers that self-associate to form rigid fibers. Protein–protein interactions regulate enzymatic activity, control progression through the cell cycle, and allow the assembly of large protein complexes that carry out many closely related reactions with a common biological function. Proteins can bind to, or be integrated into, cell membranes. The ability of binding partners to induce conformational changes in proteins allows the construction of enormously complex signaling networks.[42]:830–49 As interactions between proteins are reversible and depend heavily on the availability of different groups of partner proteins to form aggregates that are capable to carry out discrete sets of function, study of the interactions between specific proteins is a key to understand important aspects of cellular function, and ultimately the properties that distinguish particular cell types.[60][61]
The best-known role of proteins in the cell is as enzymes, which catalyse chemical reactions. Enzymes are usually highly specific and accelerate only one or a few chemical reactions. Enzymes carry out most of the reactions involved in metabolism, as well as manipulating DNA in processes such as DNA replication, DNA repair, and transcription. Some enzymes act on other proteins to add or remove chemical groups in a process known as posttranslational modification. About 4,000 reactions are known to be catalysed by enzymes.[62] The rate acceleration conferred by enzymatic catalysis is often enormous—as much as 1017-fold increase in rate over the uncatalysed reaction in the case of orotate decarboxylase (78 million years without the enzyme, 18 milliseconds with the enzyme).[63]
The molecules bound and acted upon by enzymes are called substrates. Although enzymes can consist of hundreds of amino acids, it is usually only a small fraction of the residues that come in contact with the substrate, and an even smaller fraction—three to four residues on average—that are directly involved in catalysis.[64] The region of the enzyme that binds the substrate and contains the catalytic residues is known as the active site.[54]:389
Many proteins are involved in the process of cell signaling and signal transduction. Some proteins, such as insulin, are extracellular proteins that transmit a signal from the cell in which they were synthesized to other cells in distant tissues. Others are membrane proteins that act as receptors whose main function is to bind a signaling molecule and induce a biochemical response in the cell. Many receptors have a binding site exposed on the cell surface and an effector domain within the cell, which may have enzymatic activity or may undergo a conformational change detected by other proteins within the cell.[41]:251–81
Antibodies are protein components of an adaptive immune system whose main function is to bind antigens, or foreign substances in the body, and target them for destruction. Antibodies can be secreted into the extracellular environment or anchored in the membranes of specialized B cells known as plasma cells. Whereas enzymes are limited in their binding affinity for their substrates by the necessity of conducting their reaction, antibodies have no such constraints. An antibody's binding affinity to its target is extraordinarily high.[42]:275–50
Many ligand transport proteins bind particular small biomolecules and transport them to other locations in the body of a multicellular organism. These proteins must have a high binding affinity when their ligand is present in high concentrations, and release the ligand when it is present at low concentrations in the target tissues. The canonical example of a ligand-binding protein is haemoglobin, which transports oxygen from the lungs to other organs and tissues in all vertebrates and has close homologs in every biological kingdom.[42]:222–29Lectins are sugar-binding proteins which are highly specific for their sugar moieties. Lectins typically play a role in biological recognition phenomena involving cells and proteins.[66]Receptors and hormones are highly specific binding proteins.
Transmembrane proteins can serve as ligand transport proteins that alter the permeability of the cell membrane to small molecules and ions. The membrane alone has a hydrophobic core through which polar or charged molecules cannot diffuse. Membrane proteins contain internal channels that allow such molecules to enter and exit the cell. Many ion channel proteins are specialized to select for only a particular ion; for example, potassium and sodium channels often discriminate for only one of the two ions.[41]:232–34
Other proteins that serve structural functions are motor proteins such as myosin, kinesin, and dynein, which are capable of generating mechanical forces. These proteins are crucial for cellular motility of single celled organisms and the sperm of many multicellular organisms which reproduce sexually. They generate the forces exerted by contracting muscles[42]:258–64,272 and play essential roles in intracellular transport.[54]:481,490
Methods commonly used to study protein structure and function include immunohistochemistry, site-directed mutagenesis, X-ray crystallography, nuclear magnetic resonance and mass spectrometry. The activities and structures of proteins may be examined in vitro, in vivo, and in silico. In vitro studies of purified proteins in controlled environments are useful for learning how a protein carries out its function:[67] for example, enzyme kinetics studies explore the chemical mechanism of an enzyme's catalytic activity and its relative affinity for various possible substrate molecules.[68] By contrast, in vivo experiments can provide information about the physiological role of a protein in the context of a cell or even a whole organism, and can often provide more information about protein behavior in different contexts.[69]In silico studies use computational methods to study proteins.[70]
To perform in vitro analysis, a protein must be purified away from other cellular components. This process usually begins with cell lysis, in which a cell's membrane is disrupted and its internal contents released into a solution known as a crude lysate. The resulting mixture can be purified using ultracentrifugation, which fractionates the various cellular components into fractions containing soluble proteins; membrane lipids and proteins; cellular organelles, and nucleic acids. Precipitation by a method known as salting out can concentrate the proteins from this lysate. Various types of chromatography are then used to isolate the protein or proteins of interest based on properties such as molecular weight, net charge and binding affinity.[36]:21–24 The level of purification can be monitored using various types of gel electrophoresis if the desired protein's molecular weight and isoelectric point are known, by spectroscopy if the protein has distinguishable spectroscopic features, or by enzyme assays if the protein has enzymatic activity. Additionally, proteins can be isolated according to their charge using electrofocusing.[72]
For natural proteins, a series of purification steps may be necessary to obtain protein sufficiently pure for laboratory applications. To simplify this process, genetic engineering is often used to add chemical features to proteins that make them easier to purify without affecting their structure or activity. Here, a "tag" consisting of a specific amino acid sequence, often a series of histidine residues (a "His-tag"), is attached to one terminus of the protein. As a result, when the lysate is passed over a chromatography column containing nickel, the histidine residues ligate the nickel and attach to the column while the untagged components of the lysate pass unimpeded. A number of tags have been developed to help researchers purify specific proteins from complex mixtures.[71]
The study of proteins in vivo is often concerned with the synthesis and localization of the protein within the cell. Although many intracellular proteins are synthesized in the cytoplasm and membrane-bound or secreted proteins in the endoplasmic reticulum, the specifics of how proteins are targeted to specific organelles or cellular structures is often unclear. A useful technique for assessing cellular localization uses genetic engineering to express in a cell a fusion protein or chimera consisting of the natural protein of interest linked to a "reporter" such as green fluorescent protein (GFP).[73] The fused protein's position within the cell can then be cleanly and efficiently visualized using microscopy.[74]
Other methods for elucidating the cellular location of proteins requires the use of known compartmental markers for regions such as the ER, the Golgi, lysosomes or vacuoles, mitochondria, chloroplasts, plasma membrane, etc. With the use of fluorescently tagged versions of these markers or of antibodies to known markers, it becomes much simpler to identify the localization of a protein of interest. For example, indirect immunofluorescence will allow for fluorescence colocalization and demonstration of location. Fluorescent dyes are used to label cellular compartments for a similar purpose.[75]
Other possibilities exist, as well. For example, immunohistochemistry usually uses an antibody to one or more proteins of interest that are conjugated to enzymes yielding either luminescent or chromogenic signals that can be compared between samples, allowing for localization information.[76] Another applicable technique is cofractionation in sucrose (or other material) gradients using isopycnic centrifugation.[77] While this technique does not prove colocalization of a compartment of known density and the protein of interest, it indicates an increased likelihood.[77]
Finally, the gold-standard method of cellular localization is immunoelectron microscopy. This technique uses an antibody to the protein of interest, along with classical electron microscopy techniques. The sample is prepared for normal electron microscopic examination, and then treated with an antibody to the protein of interest that is conjugated to an extremely electro-dense material, usually gold. This allows for the localization of both ultrastructural details as well as the protein of interest.[78]
Through another genetic engineering application known as site-directed mutagenesis, researchers can alter the protein sequence and hence its structure, cellular localization, and susceptibility to regulation. This technique even allows the incorporation of unnatural amino acids into proteins, using modified tRNAs,[79] and may allow the rational design of new proteins with novel properties.[80]
The total complement of proteins present at a time in a cell or cell type is known as its proteome, and the study of such large-scale data sets defines the field of proteomics, named by analogy to the related field of genomics. Key experimental techniques in proteomics include 2D electrophoresis,[81] which allows the separation of many proteins, mass spectrometry,[82] which allows rapid high-throughput identification of proteins and sequencing of peptides (most often after in-gel digestion), protein microarrays, which allow the detection of the relative levels of the various proteins present in a cell, and two-hybrid screening, which allows the systematic exploration of protein–protein interactions.[83] The total complement of biologically possible such interactions is known as the interactome.[84] A systematic attempt to determine the structures of proteins representing every possible fold is known as structural genomics.[85]
Structure determination
Discovering the tertiary structure of a protein, or the quaternary structure of its complexes, can provide important clues about how the protein performs its function and how it can be affected, i.e. in drug design. As proteins are too small to be seen under a light microscope, other methods have to be employed to determine their structure. Common experimental methods include X-ray crystallography and NMR spectroscopy, both of which can produce structural information at atomic resolution. However, NMR experiments are able to provide information from which a subset of distances between pairs of atoms can be estimated, and the final possible conformations for a protein are determined by solving a distance geometry problem. Dual polarisation interferometry is a quantitative analytical method for measuring the overall protein conformation and conformational changes due to interactions or other stimulus. Circular dichroism is another laboratory technique for determining internal β-sheet / α-helical composition of proteins. Cryoelectron microscopy is used to produce lower-resolution structural information about very large protein complexes, including assembled viruses;[41]:340–41 a variant known as electron crystallography can produce high-resolution information in some cases, especially for two-dimensional crystals of membrane proteins.[86] Solved structures are usually deposited in the Protein Data Bank (PDB), a freely available resource from which structural data about thousands of proteins can be obtained in the form of Cartesian coordinates for each atom in the protein.[87]
Many more gene sequences are known than protein structures. Further, the set of solved structures is biased toward proteins that can be easily subjected to the conditions required in X-ray crystallography, one of the major structure determination methods. In particular, globular proteins are comparatively easy to crystallize in preparation for X-ray crystallography. Membrane proteins and large protein complexes, by contrast, are difficult to crystallize and are underrepresented in the PDB.[88]Structural genomics initiatives have attempted to remedy these deficiencies by systematically solving representative structures of major fold classes. Protein structure prediction methods attempt to provide a means of generating a plausible structure for proteins whose structures have not been experimentally determined.[89]
Constituent amino-acids can be analyzed to predict secondary, tertiary and quaternary protein structure, in this case hemoglobin containing heme units
Complementary to the field of structural genomics, protein structure prediction develops efficient mathematical models of proteins to computationally predict the molecular formations in theory, instead of detecting structures with laboratory observation.[90] The most successful type of structure prediction, known as homology modeling, relies on the existence of a "template" structure with sequence similarity to the protein being modeled; structural genomics' goal is to provide sufficient representation in solved structures to model most of those that remain.[91] Although producing accurate models remains a challenge when only distantly related template structures are available, it has been suggested that sequence alignment is the bottleneck in this process, as quite accurate models can be produced if a "perfect" sequence alignment is known.[92] Many structure prediction methods have served to inform the emerging field of protein engineering, in which novel protein folds have already been designed.[93] Many proteins (in eukaryotes ~33%) contain large unstructured but biologically functional segments and can be classified as intrinsically disordered proteins. Predicting and analysing protein disorder is an important part of protein structure characterisation.[94]
Beyond classical molecular dynamics, quantum dynamics methods allow the simulation of proteins in atomistic detail with an accurate description of quantum mechanical effects. Examples include the multi-layer multi-configuration time-dependent Hartree method and the hierarchical equations of motion approach, which have been applied to plant cryptochromes[99] and bacteria light-harvesting complexes,[100] respectively. Both quantum and classical mechanical simulations of biological-scale systems are extremely computationally demanding, so distributed computing initiatives such as the Folding@home project facilitate the molecular modeling by exploiting advances in GPU parallel processing and Monte Carlo techniques.[101][102]
The total nitrogen content of organic matter is mainly formed by the amino groups in proteins. The total Kjeldahl nitrogen (TKN) is a measure of nitrogen widely used in the analysis of (waste) water, soil, food, feed and organic matter in general. As the name suggests, the Kjeldahl method is applied. More sensitive methods are available.[103][104]
Hydrolysis of protein. X = HCl and heat for industrial proteolysis. X = protease for biological proteolysis
In the absence of catalysts, proteins are slow to hydrolyze.[105] The breakdown of proteins to small peptides and amino acids (proteolysis) is a step in digestion; these breakdown products are then absorbed in the small intestine.[106] The hydrolysis of proteins relies on enzymes called proteases or peptidases. Proteases, which are themselves proteins, come in several types according to the particular peptide bonds that they cleave as well as their tendency to cleave peptide bonds at the terminus of a protein (exopeptidases) vs peptide bonds at the interior of the protein (endopeptidases).[107]Pepsin is an endopeptidase in the stomach. Subsequent to the stomach, the pancreas secretes other proteases to complete the hydrolysis, these include trypsin and chymotrypsin.[108]
Protein hydrolysis is employed commercially as a means of producing amino acids from bulk sources of protein, such as blood meal, feathers, keratin. Such materials are treated with hot hydrochloric acid, which effects the hydrolysis of the peptide bonds.[109]
Mechanical properties
The mechanical properties of proteins are highly diverse and are often central to their biological function, as in the case of proteins like keratin and collagen.[110] For instance, the ability of muscle tissue to continually expand and contract is directly tied to the elastic properties of their underlying protein makeup.[111][112] Beyond fibrous proteins, the conformational dynamics of enzymes[113] and the structure of biological membranes, among other biological functions, are governed by the mechanical properties of the proteins. Outside of their biological context, the unique mechanical properties of many proteins, along with their relative sustainability when compared to synthetic polymers, have made them desirable targets for next-generation materials design.[114][115]
Young's modulus, E, is calculated as the axial stress σ over the resulting strain ε. It is a measure of the relative stiffness of a material. In the context of proteins, this stiffness often directly correlates to biological function. For example, collagen, found in connective tissue, bones, and cartilage, and keratin, found in nails, claws, and hair, have observed stiffnesses that are several orders of magnitude higher than that of elastin,[116] which is thought to give elasticity to structures such as blood vessels, pulmonary tissue, and bladder tissue, among others.[117][118] In comparison to this, globular proteins, such as Bovine Serum Albumin, which float relatively freely in the cytosol and often function as enzymes (and thus undergoing frequent conformational changes) have comparably much lower Young's moduli.[119][120]
The Young's modulus of a single protein can be found through molecular dynamics simulation. Using either atomistic force-fields, such as CHARMM or GROMOS, or coarse-grained forcefields like Martini,[121] a single protein molecule can be stretched by a uniaxial force while the resulting extension is recorded in order to calculate the strain.[122][123] Experimentally, methods such as atomic force microscopy can be used to obtain similar data.[124] The internal dynamics of proteins involve subtle elastic and plastic deformations induced by viscoelastic forces, which can be probed by nano-rheology techniques.[125]
At the macroscopic level, the Young's modulus of cross-linked protein networks can be obtained through more traditional mechanical testing. Experimentally observed values for a few proteins can be seen below.
123Reynolds JA, Tanford C (2003). Nature's Robots: A History of Proteins (Oxford Paperbacks). New York, New York: Oxford University Press. p.15. ISBN978-0-19-860694-9.
↑Bischoff TL, Voit C (1860). Die Gesetze der Ernaehrung des Pflanzenfressers durch neue Untersuchungen festgestellt (in German). Leipzig, Heidelberg: C.F. Winter'sche Verlagshandlung.
↑Kauzmann W (May 1956). "Structural factors in protein denaturation". Journal of Cellular Physiology. Supplement. 47 (Suppl 1): 113–131. doi:10.1002/jcp.1030470410. PMID13332017.
↑Kalman SM, Linderstrøm-Lang K, Ottesen M, Richards FM (February 1955). "Degradation of ribonuclease by subtilisin". Biochimica et Biophysica Acta. 16 (2): 297–299. doi:10.1016/0006-3002(55)90224-9. PMID14363272.
↑Keskin O, Tuncbag N, Gursoy A (April 2008). "Characterization and prediction of protein interfaces to infer protein-protein interaction networks". Current Pharmaceutical Biotechnology. 9 (2): 67–76. doi:10.2174/138920108783955191. hdl:11511/32640. PMID18393863.
↑Ekman D, Björklund AK, Frey-Skött J, Elofsson A (April 2005). "Multi-domain proteins in the three kingdoms of life: orphan domains and other unassigned regions". Journal of Molecular Biology. 348 (1): 231–243. Bibcode:2005JMBio.348..231E. doi:10.1016/j.jmb.2005.02.007. PMID15808866.
↑Nelson DL, Cox MM (2005). Lehninger's Principles of Biochemistry (4thed.). New York, New York: W. H. Freeman and Company. Bibcode:2005lpbc.book.....N.
↑Gutteridge A, Thornton JM (November 2005). "Understanding nature's catalytic toolkit". Trends in Biochemical Sciences. 30 (11): 622–629. doi:10.1016/j.tibs.2005.09.006. PMID16214343.
1234567Murray RF, Harper HW, Granner DK, Mayes PA, Rodwell VW (2006). Harper's Illustrated Biochemistry. New York: Lange Medical Books/McGraw-Hill. ISBN978-0-07-146197-9.
12Reusch W (5 May 2013). "Peptides & Proteins". Michigan State University Department of Chemistry.
123Lodish H, Berk A, Matsudaira P, Kaiser CA, Krieger M, Scott MP, etal. (2004). Molecular Cell Biology (5thed.). New York, New York: WH Freeman and Company.
↑Ellis R (1979). "The most abundant protein in the world". Trends in Biochemical Sciences. 4 (11): 241–244. doi:10.1016/0968-0004(79)90212-3.
↑Dobson CM (2000). "The nature and significance of protein folding". In Pain RH (ed.). Mechanisms of Protein Folding. Oxford, Oxfordshire: Oxford University Press. pp.1–28. ISBN978-0-19-963789-8.
↑Fulton AB, Isaacs WB (April 1991). "Titin, a huge, elastic sarcomeric protein with a probable role in morphogenesis". BioEssays. 13 (4): 157–161. doi:10.1002/bies.950130403. PMID1859393. S2CID20237314.
↑Bruckdorfer T, Marder O, Albericio F (February 2004). "From production of peptides in milligram amounts for research to multi-tons quantities for drugs of the future". Current Pharmaceutical Biotechnology. 5 (1): 29–43. doi:10.2174/1389201043489620. PMID14965208.
↑Schwarzer D, Cole PA (December 2005). "Protein semisynthesis and expressed protein ligation: chasing a protein's tail". Current Opinion in Chemical Biology. 9 (6): 561–569. doi:10.1016/j.cbpa.2005.09.018. PMID16226484.
↑Pickel B, Schaller A (October 2013). "Dirigent proteins: molecular characteristics and potential biotechnological applications". Applied Microbiology and Biotechnology. 97 (19): 8427–8438. doi:10.1007/s00253-013-5167-4. PMID23989917. S2CID1896003.
↑Rüdiger H, Siebert HC, Solís D, Jiménez-Barbero J, Romero A, von der Lieth CW, etal. (April 2000). "Medicinal chemistry based on the sugar code: fundamentals of lectinology and experimental strategies with lectins as targets". Current Medicinal Chemistry. 7 (4): 389–416. doi:10.2174/0929867003375164. PMID10702616.
↑National Research Council (US) Subcommittee on Reproductive and Developmental Toxicity (2001), "Experimental Animal and In Vitro Study Designs", Evaluating Chemical and Other Agent Exposures for Reproductive and Developmental Toxicity, National Academies Press, retrieved 2024-12-23
12Terpe K (January 2003). "Overview of tag protein fusions: from molecular and biochemical fundamentals to commercial systems". Applied Microbiology and Biotechnology. 60 (5): 523–533. doi:10.1007/s00253-002-1158-6. PMID12536251. S2CID206934268.
↑Hey J, Posch A, Cohen A, Liu N, Harbers A (2008). "Fractionation of Complex Protein Mixtures by Liquid-Phase Isoelectric Focusing". 2D PAGE: Sample Preparation and Fractionation. Methods in Molecular Biology. Vol.424. pp.225–239. doi:10.1007/978-1-60327-064-9_19. ISBN978-1-58829-722-8. PMID18369866.
↑Margolin W (January 2000). "Green fluorescent protein as a reporter for macromolecular localization in bacterial cells". Methods. 20 (1): 62–72. doi:10.1006/meth.1999.0906. PMID10610805.
12Walker JH, Wilson K (2000). Principles and Techniques of Practical Biochemistry. Cambridge, UK: Cambridge University Press. pp.287–89. ISBN978-0-521-65873-7.
↑Hohsaka T, Sisido M (December 2002). "Incorporation of non-natural amino acids into proteins". Current Opinion in Chemical Biology. 6 (6): 809–815. doi:10.1016/S1367-5931(02)00376-9. PMID12470735.
↑Cedrone F, Ménez A, Quéméneur E (August 2000). "Tailoring new enzyme functions by rational redesign". Current Opinion in Structural Biology. 10 (4): 405–410. doi:10.1016/S0959-440X(00)00106-8. PMID10981626.
↑Conrotto P, Souchelnytskyi S (September 2008). "Proteomic approaches in biological and medical sciences: principles and applications". Experimental Oncology. 30 (3): 171–180. PMID18806738.
↑Koegl M, Uetz P (December 2007). "Improving yeast two-hybrid screening systems". Briefings in Functional Genomics & Proteomics. 6 (4): 302–312. doi:10.1093/bfgp/elm035. PMID18218650.
↑Hoffmann M, Wanko M, Strodel P, König PH, Frauenheim T, Schulten K, etal. (August 2006). "Color tuning in rhodopsins: the mechanism for the spectral shift between bacteriorhodopsin and sensory rhodopsin II". Journal of the American Chemical Society. 128 (33): 10808–10818. Bibcode:2006JAChS.12810808H. doi:10.1021/ja062082i. PMID16910676.
↑Mendive-Tapia D, Mangaud E, Firmino T, de la Lande A, Desouter-Lecomte M, Meyer HD, etal. (January 2018). "Multidimensional Quantum Mechanical Modeling of Electron Transfer and Electronic Coherence in Plant Cryptochromes: The Role of Initial Bath Conditions". The Journal of Physical Chemistry B. 122 (1): 126–136. Bibcode:2018JPCB..122..126M. doi:10.1021/acs.jpcb.7b10412. PMID29216421.
↑Zheng S, Javidpour L, Sahimi M, Shing KS, Nakano A (2020). "sDMD: An open source program for discontinuous molecular dynamics simulation of protein folding and aggregation". Computer Physics Communications. 247 106873. Bibcode:2020CoPhC.24706873Z. doi:10.1016/j.cpc.2019.106873.
↑Martin PD, Malley DF, Manning G, Fuller L (November 2002). "Determination of soil organic carbon and nitrogen at the field level using near-infrared spectroscopy". Canadian Journal of Soil Science. 82 (4): 413–422. Bibcode:2002CaJSS..82..413M. doi:10.4141/S01-054.
↑Radzicka A, Wolfenden R (1 January 1996). "Rates of Uncatalyzed Peptide Bond Hydrolysis in Neutral Solution and the Transition State Affinities of Proteases". Journal of the American Chemical Society. 118 (26): 6105–6109. Bibcode:1996JAChS.118.6105R. doi:10.1021/ja954077c.
↑Switzar L, Giera M, Niessen WM (1 March 2013). "Protein Digestion: An Overview of the Available Techniques and Recent Developments". Journal of Proteome Research. 12 (3): 1067–1077. doi:10.1021/pr301201x. PMID23368288.
↑Debelle L, Tamburro AM (February 1999). "Elastin: molecular description and function". The International Journal of Biochemistry & Cell Biology. 31 (2): 261–272. doi:10.1016/S1357-2725(98)00098-3. PMID10216959.
↑Tan R, Shin J, Heo J, Cole BD, Hong J, Jang Y (October 2020). "Tuning the Structural Integrity and Mechanical Properties of Globular Protein Vesicles by Blending Crosslinkable and NonCrosslinkable Building Blocks". Biomacromolecules. 21 (10): 4336–4344. doi:10.1021/acs.biomac.0c01147. PMID32955862.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.