Super-resolution microscopy is a series of techniques in optical microscopy that allow such images to have resolutions higher than those imposed by the diffraction limit,[1][2] which is due to the diffraction of light.[3] Super-resolution imaging techniques rely on the near-field (photon-tunneling microscopy[4] as well as those that use the Pendry Superlens and near field scanning optical microscopy) or on the far-field. Among techniques that rely on the latter are those that improve the resolution only modestly (up to about a factor of two) beyond the diffraction-limit, such as confocal microscopy with closed pinhole or aided by computational methods such as deconvolution[5] or detector-based pixel reassignment (e.g. re-scan microscopy,[6] pixel reassignment[7]), the 4Pi microscope, and structured-illumination microscopy technologies such as SIM[8][9] and SMI.
There are two major groups of methods for super-resolution microscopy in the far-field that can improve the resolution by a much larger factor:[10]
Deterministic super-resolution: the most commonly used emitters in biological microscopy, fluorophores, show a nonlinear response to excitation, which can be exploited to enhance resolution. Such methods include STED, GSD, RESOLFT and SSIM.
Stochastic super-resolution: the chemical complexity of many molecular light sources gives them a complex temporal behavior, which can be used to make several nearby fluorophores emit light at separate times and thereby become resolvable in time. These methods include super-resolution optical fluctuation imaging (SOFI) and all single-molecule localization methods (SMLM), such as SPDM, SPDMphymod, PALM, FPALM, STORM, and dSTORM.
By 1978, the first theoretical ideas had been developed to break the Abbe limit, which called for using a 4Pi microscope as a confocal laser-scanning fluorescence microscope where the light is focused from all sides to a common focus that is used to scan the object by 'point-by-point' excitation combined with 'point-by-point' detection.[14] However the publication from 1978 [15] had drawn an improper physical conclusion (i.e. a point-like spot of light) and had completely missed the axial resolution increase as the actual benefit of adding the other side of the solid angle.[16]
Some of the following information was gathered (with permission) from a chemistry blog's review of sub-diffraction microscopy techniques.[17][18]
In 1986, a super-resolution optical microscope based on stimulated emission was patented by Okhonin.[19]
Super-resolution techniques
Photon tunneling microscopy (PTM)
Photon tunneling microscopy (PTM)[20] provides real-time whole-field non-scanning imaging with sub-wavelength lateral resolution and vertical resolution as low as 1 nanometer, as demonstrated by John M. Guerra. (Note the differentiation of PTM with photon scanning tunneling microscopy (PSTM), which is essentially a re-naming of near-field scanning optical microscopy (NSOM) that exploits the near-field from a sharp optical fiber tip positioned extremely close to the sample surface, typically within a few nanometers, such that when the fiber is scanned along the sample surface an image is built up).
PTM provides sub-wavelength spatial resolution due to its capture of bound high-frequency diffracted light at the sample and conversion into image-forming propagating light. Easy insertion of the sample surface into the evanescent field at the distal end of the microscope is provided by a flexible "transducer" of a polymer or thin glass that is placed proximal to the sample.[21] This transducer is flexible enough to follow macro-curves on the sample while providing a stiff flat reference locally. The vertical resolution is limited only by the photometric resolution of the whole-field detector, and can be as low as 1 nanometer. The technique has been applied to biological and solid-state samples, offering insights into surface morphology and optical properties at the nanoscale.
Phase-shifted PTM aka Phase-shifted Evanescent field Microscopy (PEM)
In further development of this work in 1996, John M. Guerra showed that super-resolved lateral topography even higher than is achieved with PTM is attained by phase-shifting the evanescent field. In propagating light, planes of equal phase are parallel to planes of equal amplitude, such that phase-shifting produces sub-wavelength super-resolution along the vertical axis, such as achieved with interferometers produced by WYKO and ZYGO for measurement of optical surfaces. However, the evanescent field is inhomogenous in that the planes of equal amplitude are parallel to the sample surface (thus the high vertical resolution in PTM), while the planes of equal phase are perpendicular to them and to the surface. Shifting the phase therefore now can produce sub-wavelength super-resolution in the lateral axis that is even higher than that of PTM. While there are many ways to initiate this phase shift, as taught by Guerra in several U.S. patents[22] assigned to the Polaroid Corporation, the first demonstration employed a Meadowlark Optics phase retarder and polarizer combination. Subtracting consecutive images for which the phase was shifted by a small value produced resolution Licenses to this technology were procured by Dyer Energy Systems, Calimetrics Inc., and Nanoptek Corp. for use of this super-resolution technique in optical data storage and microscopy. In 2001, with Dmitri Vezenov, Guerra published[23] a fully-resolved image of 200nm spheres, close-packed hexagonally, while illuminated with 650nm light, for a resolution of 0.3 lambda. While impressive, potential resolution can be much higher, as it is limited only by the magnitude of the phase-shift.
Local enhancement / ANSOM / optical nano-antennas
Apertureless near-field scanning optical microscopy (ANSOM), also known as scattering-type scanning near-field optical microscopy (s-SNOM), achieves super-resolution imaging through the local enhancement of the optical field near a sharp tip, often made of metal. In ANSOM, a metallic or dielectric probe interacts with incident light, creating a confined and enhanced electromagnetic field at the apex of the tip due to localized surface plasmon resonance.
Optical nano-antennas can further improve resolution by acting as resonators that concentrate light into nanoscale volumes, enhancing local field strength and sensitivity. These nano-antennas are engineered to resonate at specific optical frequencies and are used both in illumination and detection modes to achieve spatial resolutions well below the diffraction limit.
Near-field optical random mapping (NORM) microscopy
Near-field optical random mapping (NORM) microscopy is a method of optical near-field acquisition by a far-field microscope through the observation of nanoparticles' Brownian motion in an immersion liquid.[24][25]
NORM uses object surface scanning by stochastically moving nanoparticles. Through the microscope, nanoparticles look like symmetric round spots. The spot width is equivalent to the point spread function (~ 250nm) and is defined by the microscope resolution. Lateral coordinates of the given particle can be evaluated with a precision much higher than the resolution of the microscope. By collecting the information from many frames one can map out the near field intensity distribution across the whole field of view of the microscope. In comparison with NSOM and ANSOM this method does not require any special equipment for tip positioning and has a large field of view and a depth of focus. Due to the large number of scanning "sensors" one can achieve image acquisition in a shorter time.
The improvement in resolution is achieved by using two opposing objective lenses, both of which are focused to the same geometric location. Also, the difference in optical path length through each of the two objective lenses is carefully minimized. By this, molecules residing in the common focal area of both objectives can be illuminated coherently from both sides, and the reflected or emitted light can be collected coherently, i.e. coherent superposition of emitted light on the detector is possible. The solid angle that is used for illumination and detection is increased and approaches the ideal case, where the sample is illuminated and detected from all sides simultaneously.[26][27]
Up to now, the best quality in a 4Pi microscope has been reached in conjunction with STED microscopy in fixed cells[28] and RESOLFT microscopy with switchable proteins in living cells.[29]
Comparison of the resolution obtained by confocal laser scanning microscopy (top) and 3D structured illumination microscopy (3D-SIM-Microscopy, bottom). Shown are details of a nuclear envelope. Nuclear pores (anti-NPC) red, nuclear envelope (anti-Lamin) green, chromatin (DAPI-staining) blue. Scale bar: 1μm.
Structured illumination microscopy (SIM) enhances spatial resolution by collecting information from frequency space outside the observable region. This process is done in reciprocal space: the Fourier transform (FT) of an SI image contains superimposed additional information from different areas of reciprocal space; with several frames where the illumination is shifted by some phase, it is possible to computationally separate and reconstruct the FT image, which has much more resolution information. The reverse FT returns the reconstructed image to a super-resolution image.
SIM could potentially replace electron microscopy as a tool for some medical diagnoses. These include diagnosis of kidney disorders,[30] kidney cancer,[31] and blood diseases.[32]
Although the term "structured illumination microscopy" was coined by others in later years, John M. Guerra (1995) first published results[33] in which light patterned by a 50nm pitch grating illuminated a second grating of pitch 50nm, with the gratings rotated with respect to each other by the angular amount needed to achieve magnification. Although the illuminating wavelength was 650nm, the 50nm grating was easily resolved. This showed a nearly 5-fold improvement over the Abbe resolution limit of 232nm that should have been the smallest obtained for the numerical aperture and wavelength used.
One implementation of structured illumination is known as spatially modulated illumination (SMI). Like standard structured illumination, the SMI technique modifies the point spread function (PSF) of a microscope in a suitable manner. In this case however, "the optical resolution itself is not enhanced";[34] instead structured illumination is used to maximize the precision of distance measurements of fluorescent objects, to "enable size measurements at molecular dimensions of a few tens of nanometers".[34]
The Vertico SMI microscope achieves structured illumination by using one or two opposing interfering laser beams along the axis. The object being imaged is then moved in high-precision steps through the wave field, or the wave field itself is moved relative to the object by phase shifts. This results in an improved axial size and distance resolution.[34][35][36]
SMI can be combined with other super resolution technologies, for instance with 3D LIMON or LSI-TIRF as a total internal reflection interferometer with laterally structured illumination (this last instrument and technique is essentially a phase-shifted photon tunneling microscope, which employs a total internal reflection light microscope with phase-shifted evanescent field (John M. Guerra, 1996).[22] This SMI technique allows one to acquire light-optical images of autofluorophore distributions in sections from human eye tissue with previously unmatched optical resolution. Use of three different excitation wavelengths (488, 568, and 647nm), enables one to gather spectral information about the autofluorescence signal. This has been used to examine human eye tissue affected by macular degeneration.[37]
Biosensing
Biosensing is crucial for understanding the activities of cellular components in cell biology. Genetically encoded sensors have transformed this field and typically consist of two parts: the sensing domain, which detects cellular activity or interactions, and the reporting domain, which produces measurable signals. There are two main types of sensors: FRET-based sensors using two fluorophores for precise quantification but with some limitations, and single-fluorophore biosensors that are smaller, faster, and allow for multiplexed experiments, but may have challenges in obtaining absolute values and detecting response saturation. Various microscopy methods, including super-resolution optical fluctuation imaging, have been used to quantify and monitor biological activities in real time. Examples include calcium, pH, and voltage sensing. Greenwald et al. offer a more comprehensive overview of these applications.[38]
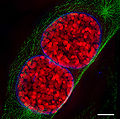
Resolution improvement between traditional confocal microscopy and STED microscopy.
Stimulated emission depletion microscopy (STED microscopy) uses two laser pulses, the excitation pulse for excitation of the fluorophores to their fluorescent state and the STED pulse for the de-excitation of fluorophores by means of stimulated emission.[19][43][44][45][46][47] In practice, the excitation laser pulse is first applied whereupon a STED pulse soon follows (STED without pulses using continuous wave lasers is also used). Furthermore, the STED pulse is modified in such a way so that it features a zero-intensity spot that coincides with the excitation focal spot. Due to the non-linear dependence of the stimulated emission rate on the intensity of the STED beam, all the fluorophores around the focal excitation spot will be in their off state (the ground state of the fluorophores). By scanning this focal spot, one retrieves the image. The full width at half maximum (FWHM) of the point spread function (PSF) of the excitation focal spot can theoretically be compressed to an arbitrary width by raising the intensity of the STED pulse, according to equation (1).
(1)
where ∆r is the lateral resolution, ∆ is the FWHM of the diffraction limited PSF, Imax is the peak intensity of the STED laser, and is the threshold intensity needed in order to achieve saturated emission depletion.
The main disadvantage of STED, which has prevented its widespread use, is that the machinery is complicated. On the one hand, the image acquisition speed is relatively slow for large fields of view because of the need to scan the sample in order to retrieve an image. On the other hand, it can be very fast for smaller fields of view: recordings of up to 80 frames per second have been shown.[48][49] Due to a large Is value associated with STED, there is the need for a high-intensity excitation pulse, which may cause damage to the sample.
Ground state depletion (GSD)
Ground state depletion microscopy (GSD microscopy) uses the triplet state of a fluorophore as the off-state and the singlet state as the on-state, whereby an excitation laser is used to drive the fluorophores at the periphery of the singlet state molecule to the triplet state. This is much like STED, where the off-state is the ground state of fluorophores, which is why equation (1) also applies in this case. The value is smaller than in STED, making super-resolution imaging possible at a much smaller laser intensity. Compared to STED, though, the fluorophores used in GSD are generally less photostable; and the saturation of the triplet state may be harder to realize.[50]
Saturated structured-illumination microscopy (SSIM) exploits the nonlinear dependence of the emission rate of fluorophores on the intensity of the excitation laser.[51] By applying a sinusoidal illumination pattern[52] with a peak intensity close to that needed in order to saturate the fluorophores in their fluorescent state, one retrieves Moiré fringes. The fringes contain high order spatial information that may be extracted by computational techniques. Once the information is extracted, a super-resolution image is retrieved.
SSIM requires shifting the illumination pattern multiple times, effectively limiting the temporal resolution of the technique. In addition there is the need for very photostable fluorophores, due to the saturating conditions, which inflict radiation damage on the sample and restrict the possible applications for which SSIM may be used.
Examples of this microscopy are shown under section Structured illumination microscopy (SIM): images of cell nuclei and mitotic stages recorded with 3D-SIM Microscopy.
Stochastic functional techniques
Localization microscopy
Single-molecule localization microscopy (SMLM) summarizes all microscopical techniques that achieve super-resolution by isolating emitters and fitting their images with the point spread function (PSF). Normally, the width of the point spread function (~ 250nm) limits resolution. However, given an isolated emitter, one is able to determine its location with a precision only limited by its intensity according to equation (2).[53]
(2)
where Δloc is the localization precision, Δ is the FWHM (full width at half maximum) of the PSF and N is the number of collected photons.
This fitting process can only be performed reliably for isolated emitters (see Deconvolution), and interesting biological samples are so densely labeled with emitters that fitting is impossible when all emitters are active at the same time. SMLM techniques solve this dilemma by activating only a sparse subset of emitters at the same time, localizing these few emitters very precisely, deactivating them and activating another subset.
Considering background and camera pixelation, and using Gaussian approximation for the point spread function (Airy disk) of a typical microscope, the theoretical resolution is proposed by Thompson et al.[54] and fine-tuned by Mortensen et al.:[55]
where
* σ is the Gaussian standard deviation of the center locations of the same molecule if measured multiple times (e.g. frames of a video). (unit m)
* σPSF is the Gaussian standard deviation of the point spread function, whose FWHM following the Ernst Abbe equation d = λ/(2 N.A.). (unit m)
* a is the size of each image pixel. (unit m)
* Nsig is the photon counts of the total PSF over all pixels of interest. (unitless)
* Nbg the average background photon counts per pixel (dark counts already removed), which is approximated to be the square of the Gaussian standard deviation of the Poisson distribution background noise of each pixel over time or standard deviation of all pixels with background noise only, σbg2. The larger the σbg2, the better the approximation (e.g. good for σbg2 >10, excellent for σbg2 >1000). (unitless)
Generally, localization microscopy is performed with fluorophores. Suitable fluorophores (e.g. for STORM) reside in a non-fluorescent dark state for most of the time and are activated stochastically, typically with an excitation laser of low intensity. A readout laser stimulates fluorescence and bleaches or photoswitches the fluorophores back to a dark state, typically within 10–100 ms. In Points Accumulation for Imaging in Nanoscale Topography (PAINT), the fluorophores are nonfluorescent before binding and afterward become fluorescent. The photons emitted during the fluorescent phase are collected with a camera and the resulting image of the fluorophore (which is distorted by the PSF) can be fitted with very high precision, even on the order of a few Angstroms.[56] Repeating the process several thousand times ensures that all fluorophores can go through the bright state and are recorded. A computer then reconstructs a super-resolved image.
The desirable traits of fluorophores used for these methods, in order to maximize the resolution, are that they should be bright. That is, they should have a high extinction coefficient and a high quantum yield. They should also possess a high contrast ratio (ratio between the number of photons emitted in the light state and the number of photons emitted in the dark state). Also, a densely labeled sample is desirable, according to the Nyquist criteria.
The multitude of localization microscopy methods differ mostly in the type of fluorophores used.
Stochastic optical reconstruction microscopy (STORM), photo activated localization microscopy (PALM), and fluorescence photo-activation localization microscopy (FPALM) are super-resolution imaging techniques that use sequential activation and time-resolved localization of photoswitchable fluorophores to create high resolution images. During imaging, only an optically resolvable subset of fluorophores is activated to a fluorescent state at any given moment, such that the position of each fluorophore can be determined with high precision by finding the centroid positions of the single-molecule images of a particular fluorophore. One subset of fluorophores is subsequently deactivated, and another subset is activated and imaged. Iteration of this process allows numerous fluorophores to be localized and a super-resolution image to be constructed from the image data.
These three methods were published independently over a short period of time, and their principles are identical. STORM was originally described using Cy5 and Cy3 dyes attached to nucleic acids or proteins,[57] while PALM and FPALM were described using photoswitchable fluorescent proteins.[58][59] In principle any photoswitchable fluorophore can be used, and STORM has been demonstrated with a variety of different probes and labeling strategies. Using stochastic photoswitching of single fluorophores, such as Cy5,[60] STORM can be performed with a single red laser excitation source. The red laser both switches the Cy5 fluorophore to a dark state by formation of an adduct[61][62] and subsequently returns the molecule to the fluorescent state. Many other dyes have been also used with STORM.[63][64][65][66][67][68]
In addition to single fluorophores, dye-pairs consisting of an activator fluorophore (such as Alexa 405, Cy2, or Cy3) and a photoswitchable reporter dye (such as Cy5, Alexa 647, Cy5.5, or Cy7) can be used with STORM.[57][69][70] In this scheme, the activator fluorophore, when excited near its absorption maximum, serves to reactivate the photoswitchable dye to the fluorescent state. Multicolor imaging has been performed by using different activation wavelengths to distinguish dye-pairs, depending on the activator fluorophore used,[69][70][71] or using spectrally distinct photoswitchable fluorophores, either with or without activator fluorophores.[63][72][73] Photoswitchable fluorescent proteins can be used as well.[58][59][73][74] Highly specific labeling of biological structures with photoswitchable probes has been achieved with antibody staining,[69][70][71][75] direct conjugation of proteins,[76] and genetic encoding.[58][59][73][74]
STORM has also been extended to three-dimensional imaging using optical astigmatism, in which the elliptical shape of the point spread function encodes the x, y, and z positions for samples up to several micrometers thick,[70][75] and has been demonstrated in living cells.[73] To date, the spatial resolution achieved by this technique is ~20nm in the lateral dimensions and ~50nm in the axial dimension; and the temporal resolution is as fast as 0.1–0.33s.[citation needed]
Spectral precision distance microscopy (SPDM)
Single YFP molecule super resolution microscopy / SPDMphymod
A single, tiny source of light can be located much better than the resolution of a microscope usually allows for: although the light will produce a blurry spot, computer algorithms can be used to accurately calculate the center of the blurry spot, taking into account the point spread function of the microscope, the noise properties of the detector, etc. However, this approach does not work when there are too many sources close to each other: the sources then all blur together.
Spectral precision distance microscopy (SPDM) is a family of localizing techniques in fluorescence microscopy which gets around the problem of there being many sources by measuring just a few sources at a time, so that each source is "optically isolated" from the others (i.e., separated by more than the microscope's resolution, typically ~200-250nm).[77][78][79] This "optical isolation" requires that the particles under examination have different spectral signatures, so that it is possible to look at light from just a few molecules at a time by using the appropriate light sources and filters. This achieves an effective optical resolution several times better than the conventional optical resolution that is represented by the half-width of the main maximum of the effective point image function.[77]
The structural resolution achievable using SPDM can be expressed in terms of the smallest measurable distance between two punctiform particles of different spectral characteristics ("topological resolution"). Modeling has shown that under suitable conditions regarding the precision of localization, particle density, etc., the "topological resolution" corresponds to a "space frequency" that, in terms of the classical definition, is equivalent to a much improved optical resolution. Molecules can also be distinguished in even more subtle ways based on fluorescent lifetime and other techniques.[77]
An important application is in genome research (study of the functional organization of the genome). Another important area of use is research into the structure of membranes.
SPDMphymod
Dual color localization microscopy SPDMphymod/super-resolution microscopy with GFP & RFP fusion proteins
Localization microscopy for many standard fluorescent dyes like GFP, Alexa Fluor dyes, and fluorescein molecules is possible if certain photo-physical conditions are present. With this so-called physically modifiable fluorophores (SPDMphymod) technology, a single laser wavelength of suitable intensity is sufficient for nanoimaging[80] in contrast to other localization microscopy technologies that need two laser wavelengths when special photo-switchable/photo-activatable fluorescence molecules are used. A further example of the use of SPDMphymod is an analysis of tobacco mosaic virus (TMV) particles[81] or the study of virus–cell interaction.[82][83]
Based on singlet–triplet state transitions it is crucial for SPDMphymod that this process is ongoing and leading to the effect that a single molecule comes first into a very long-living reversible dark state (with half-life of as much as several seconds) from which it returns to a fluorescent state emitting many photons for several milliseconds before it returns into a very long-living, so-called irreversible dark state. SPDMphymod microscopy uses fluorescent molecules that emit the same spectral light frequency but with different spectral signatures based on the flashing characteristics. By combining two thousands images of the same cell, it is possible, using laser optical precision measurements, to record localization images with significantly improved optical resolution.[84]
Standard fluorescent dyes already successfully used with the SPDMphymod technology are GFP, RFP, YFP, Alexa 488, Alexa 568, Alexa 647, Cy2, Cy3, Atto 488 and fluorescein.
Cryogenic optical localization in 3D (COLD)
Cryogenic Optical Localization in Three Dimensions (COLD) allows to determine the four biotin binding sites in the protein streptavidin.
Cryogenic Optical Localization in 3D (COLD) is a method that allows localizing multiple fluorescent sites within a single small- to medium-sized biomolecule with Angstrom-scale resolution.[56] The localization precision in this approach is enhanced because the slower photochemistry at low temperatures leads to a higher number of photons that can be emitted from each fluorophore before photobleaching.[85][86] Consequently, cryogenic stochastic localization microscopy achieves the sub-molecular resolution required to resolve the 3D positions of several fluorophores attached to a small protein. By employing algorithms known from electron microscopy, the 2D projections of fluorophores are reconstructed into a 3D configuration. COLD brings fluorescence microscopy to its fundamental limit, depending on the size of the label. The method can also be combined with other structural biology techniques—such as X-ray crystallography, magnetic resonance spectroscopy, and electron microscopy—to provide valuable complementary information and specificity.
Binding-activated localization microscopy (BALM)
fBALM Super-resolution single molecule localisation microscopy using DNA structure fluctuation assisted binding activated localisation microscopy
Binding-activated localization microscopy (BALM) is a general concept for single-molecule localization microscopy (SMLM): super-resolved imaging of DNA-binding dyes based on modifying the properties of DNA and a dye.[87] By careful adjustment of the chemical environment—leading to local, reversible DNA melting and hybridization control over the fluorescence signal—DNA-binding dye molecules can be introduced. Intercalating and minor-groove binding DNA dyes can be used to register and optically isolate only a few DNA-binding dye signals at a time. DNA structure fluctuation-assisted BALM (fBALM) has been used to nanoscale differences in nuclear architecture, with an anticipated structural resolution of approximately 50nm. Imaging chromatin nanostructure with binding-activated localization microscopy based on DNA structure fluctuations.[88] Recently, the significant enhancement of fluorescence quantum yield of NIAD-4 upon binding to an amyloid was exploited for BALM imaging of amyloid fibrils[89] and oligomers.[90]
Points accumulation for imaging in nanoscale topography (PAINT)
Points accumulation for imaging in nanoscale topography (PAINT) is a single-molecule localization method that achieves stochastic single-molecule fluorescence by molecular adsorption/absorption and photobleaching/desorption.[91][92] The first dye used was Nile red which is nonfluorescent in aqueous solution but fluorescent when inserted into a hydrophobic environment, such as micelles or living cell walls. Thus, the concentration of the dye is kept small, at the nanomolar level, so that the molecule's sorption rate to the diffraction-limited area is in the millisecond region. The stochastic binding of single-dye molecules (probes) to an immobilized target can be spatially and temporally resolved under a typical widefield fluorescence microscope. Each dye is photobleached to return the field to a dark state, so the next dye can bind and be observed. The advantage of this method, compared to other stochastic methods, is that in addition to obtaining the super-resolved image of the fixed target, it can measure the dynamic binding kinetics of the diffusing probe molecules, in solution, to the target.[93][92]
Combining 3D super-resolution technique (e.g. the double-helix point spread function develop in Moerner's group), photo-activated dyes, power-dependent active intermittency, and points accumulation for imaging in nanoscale topography, SPRAIPAINT (SPRAI=Super resolution by PoweR-dependent Active Intermittency[94]) can super-resolve live-cell walls.[95] PAINT works by maintaining a balance between the dye adsorption/absorption and photobleaching/desorption rates. This balance can be estimated with statistical principles.[96] The adsorption or absorption rate of a dilute solute to a surface or interface in a gas or liquid solution can be calculated using Fick's laws of diffusion. The photobleaching/desorption rate can be measured for a given solution condition and illumination power density.
DNA-PAINT has been further extended to use regular dyes, where the dynamic binding and unbinding of a dye-labeled DNA probe to a fixed DNA origami is used to achieve stochastic single-molecule imaging.[97][98] DNA-PAINT is no longer limited to environment-sensitive dyes and can measure both the adsorption and the desorption kinetics of the probes to the target. The method uses the camera blurring effect of moving dyes. When a regular dye is diffusing in the solution, its image on a typical CCD camera is blurred because of its relatively fast speed and the relatively long camera exposure time, contributing to the fluorescence background. However, when it binds to a fixed target, the dye stops moving; and clear input into the point spread function can be achieved.
The term for this method is mbPAINT ("mb" standing for motion blur).[99] When a total internal reflection fluorescence microscope (TIRF) is used for imaging, the excitation depth is limited to ~100nm from the substrate, which further reduces the fluorescence background from the blurred dyes near the substrate and the background in the bulk solution. Very bright dyes can be used for mbPAINT which gives typical single-frame spatial resolutions of ~20nm and single-molecule kinetic temporal resolutions of ~20ms under relatively mild photoexcitation intensities, which is useful in studying molecular separation of single proteins.[100]
By using a secondary DNA strand that couples to the primary (antibody-conjugated) strand, the fluorescent label can be gently stripped, allowing multiplexed localization of 30 different proteins. This method, called SUM-PAINT, has been used to map the localization of synaptic proteins at 5 nm resolution, revealing differences in the architecture of excitatory, inhibitory and mixed synapses.[101]
The temporal resolution has been further improved (20 times) using a rotational phase mask placed in the Fourier plane during data acquisition and resolving the distorted point spread function that contains temporal information. The method was named Super Temporal-Resolved Microscopy (STReM).[102]
Optical resolution of cellular structures in the range of about 50nm can be achieved, even in label-free cells, using localization microscopy SPDM.
By using two different laser wavelengths, SPDM reveals cellular objects which are not detectable under conventional fluorescence wide-field imaging conditions, beside making for a substantial resolution improvement of autofluorescent structures.
As a control, the positions of the detected objects in the localization image match those in the bright-field image.[103]
Label-free superresolution microscopy has also been demonstrated using the fluctuations of a surface-enhanced Raman scattering signal on a highly uniform plasmonic metasurface.[104]
Direct stochastical optical reconstruction microscopy (dSTORM)
dSTORM uses the photoswitching of a single fluorophore. In dSTORM, fluorophores are embedded in a reducing and oxidizing buffering system (ROXS) and fluorescence is excited. Sometimes, stochastically, the fluorophore will enter a triplet or some other dark state that is sensitive to the oxidation state of the buffer, from which they can be made to fluoresce, so that single molecule positions can be recorded.[105] Development of the dSTORM method occurred at 3 independent laboratories at about the same time and was also called "reversible photobleaching microscopy" (RPM),[106] "ground state depletion microscopy followed by individual molecule return" (GSDIM),[107] as well as the now generally accepted moniker dSTORM.[108]
Software for localization microscopy
Localization microscopy depends heavily on software that can precisely fit the point spread function (PSF) to millions of images of active fluorophores within a few minutes.[109] Since the classical analysis methods and software suites used in the natural sciences are too slow to computationally solve these problems, often taking hours of computation for processing data measured in minutes, specialised software programs have been developed. Many of these localization software packages are open-source; they are listed at SMLM Software Benchmark.[110] Once molecule positions have been determined, the locations need to be displayed and several algorithms for display have been developed.[111]
Random Illumination Microscopy (RIM)
Random Illumination Microscopy (RIM) is a super-resolution imaging technique that employs random or pseudo-random wide-field illuminations generated by a laser. This method enables the reconstruction of a high-resolution image from multiple low-resolution frames captured under varying, unknown illumination patterns, achieving resolutions down to 90 nanometers. RIM is particularly advantageous for imaging thick, living samples due to its minimal phototoxicity and robust z-sectioning capabilities. Additionally, its resistance to optical aberrations makes it a highly effective tool for biological research.
It is possible to circumvent the need for PSF fitting inherent in single molecule localization microscopy (SMLM) by directly computing the temporal autocorrelation of pixels. This technique is called super-resolution optical fluctuation imaging (SOFI) and has been shown to be more precise than SMLM when the density of concurrently active fluorophores is very high.
Omnipresent Localization Microscopy (OLM)
Omnipresent Localisation Microscopy (OLM) is an extension of Single Molecule Microscopy (SMLM) techniques that allow high-density single molecule imaging with an incoherent light source (such as a mercury-arc lamp) and a conventional epifluorescence microscope setup.[112] A short burst of deep-blue excitation (with a 350-380nm, instead of a 405nm, laser) enables a prolonged reactivation of molecules, for a resolution of 90nm on test specimens. Finally, correlative STED and SMLM imaging can be performed on the same biological sample using a simple imaging medium, which can provide a basis for a further enhanced resolution. These findings can democratize super-resolution imaging and help any scientist to generate high-density single-molecule images even with a limited budget.
Resolution Enhancement by Sequential Imaging (RESI)
Resolution enhancement by sequential imaging (RESI) is an extension of DNA-PAINT that can achieve theoretically unlimited resolution.[113] Rather than using one label type to identify a given target species, copies of the same target are labeled with orthogonal DNA sequences. Upon sequential (i.e. separated) imaging, localization clouds that would overlap in conventional SMLM can be (1) resolved and (2) combined into a single "super" localization, the precision of which scales with the underlying number of localizations. As the number of achievable localizations in DNA-PAINT is unlimited, so is the theoretical resolution of RESI. Overlaying the RESI localizations from the underlying imaging rounds creates a composite, highly resolved image.
Combination of techniques
3D light microscopical nanosizing (LIMON) microscopy
3D Dual Colour Super Resolution Microscopy with Her2 and Her3 in breast cells, standard dyes: Alexa 488, Alexa 568 LIMON
Light MicrOscopical Nanosizing microscopy (3D LIMON) images, using the Vertico SMI microscope, are made possible by the combination of SMI and SPDM, whereby first the SMI, and then the SPDM, process is applied.
The SMI process determines the center of particles and their spread in the direction of the microscope axis. While the center of particles/molecules can be determined with a precision of 1–2nm, the spread around this point can be determined down to an axial diameter of approximately 30–40nm.
Subsequently, the lateral position of the individual particle/molecule is determined using SPDM, achieving a precision of a few nanometers.[114]
As a biological application in the 3D dual color mode, the spatial arrangements of Her2/neu and Her3 clusters was achieved. The positions in all three directions of the protein clusters could be determined with an accuracy of about 25nm.[115]
Integrated correlative light and electron microscopy
Combining a super-resolution microscope with an electron microscope enables the visualization of contextual information, with the labelling provided by fluorescence markers. This overcomes the problem of the black backdrop that the researcher is left with when using only a light microscope. In an integrated system, the sample is measured by both microscopes simultaneously.[116]
SR-FACT
SR-FACT imaging combines super resolution and assisted diffraction computational tomography. This technique has unique capabilities applicable to cell biology, particularly focused on the cell'sinteractome.[117]
Enhancing of techniques using neural networks
Recently, owing to advancements in artificial intelligence computing, deep learning neural networks (GANs) have been used for super-resolution enhancing of photographic images extracted from optical microscopes,[118] enhancing resolution from 40x to 100x.[119] Resolution increases from 20x with an optical microscope to 1500x, comparable to a scanning electron microscope, via a neural lens.[120] These techniques have applications in super-resolving images from positron-emission tomography and fluorescence microscopy.[121]
↑U.S. Pat. No. 5,666,197: Apparatus and methods employing phase control and analysis of evanescent illumination for imaging and metrology of subwavelength lateral surface topography; John M. Guerra, September 1997. Assigned to Polaroid Corp.
↑SPIE (March 2015). "W.E. Moerner plenary presentation: Single-molecule spectroscopy, imaging, and photocontrol -- foundations for super-resolution microscopy". SPIE Newsroom. doi:10.1117/2.3201503.17.
↑Cremer C, Cremer T (September 1978). "Considerations on a laser-scanning-microscope with high resolution and depth of field". Microscopica Acta. 81 (1): 31–44. PMID713859.
↑C. Cremer and T. Cremer (1978): Considerations on a laser-scanning-microscope with high resolution and depth of field Microscopica Acta VOL. 81 NUMBER 1 September, pp. 31—44 (1978)
12V.A. Okhonin, Method of investigating specimen microstructure, Patent SU 1374922, priority date 10 April 1986, Published on July 30, 1991, Soviet Patents Abstracts, Section EI, Week 9218, Derwent Publications Ltd., London, GB; Class S03, p. 4. Cited by patents US 5394268 A (1993) and US RE38307 E1 (1995). From the English translation: "The essence of the invention is as follows. Luminescence is excited in a sample placed in the field of several standing light waves, which cause luminescence quenching because of stimulated transitions...".
↑NIST ATP Funding No. 70NANB7H3054: Calimetrics East Final MORE Report, December 15, 2001, page 5.
↑US patent 2009/0116,024, priority date 7 April 2006: J. V. Mikliaev, S. A. Asselborn Method for obtaining a high resolution image
↑Miklyaev YV, Asselborn SA, Zaytsev KA, Darscht MY (2014). "Superresolution microscopy in far-field by near-field optical random mapping nanoscopy". Appl. Phys. Lett. 105 (11): 113103(1–4). Bibcode:2014ApPhL.105k3103M. doi:10.1063/1.4895922.
↑Fernández-Suárez M, Ting AY (December 2008). "Fluorescent probes for super-resolution imaging in living cells". Nature Reviews Molecular Cell Biology. 9 (12): 929–43. doi:10.1038/nrm2531. PMID19002208. S2CID2752640.
↑Sätzler B, Cremer E (1 February 1998). "High-precision distance measurements and volume-conserving segmentation of objects near and below the resolution limit in three-dimensional confocal fluorescence microscopy". Journal of Microscopy. 189 (2): 118–136. doi:10.1046/j.1365-2818.1998.00276.x. S2CID73578516.
↑Zondervan R, Kulzer F, Kolchenko M, Orrit M (2004). "Photobleaching of Rhodamine 6G in Poly(vinyl alcohol) at the Ensemble and Single-Molecule Levels". J. Phys. Chem. A. 108 (10): 1657–1665. Bibcode:2004JPCA..108.1657Z. doi:10.1021/jp037222e.
↑Weisenburger S, Jing B, Renn A, Sandoghdar V (2013). Verma P, Egner A (eds.). "Cryogenic localization of single molecules with angstrom precision". Proc. SPIE. Nanoimaging and Nanospectroscopy. 8815: 88150D. Bibcode:2013SPIE.8815E..0DW. doi:10.1117/12.2025373. S2CID120610755.
↑Huh H, Lee J, Kim HJ, Hohng S, Kim SK (2017). "Morphological analysis of oligomeric vs. fibrillar forms of α-synuclein aggregates with super-resolution BALM imaging". Chemical Physics Letters. 690: 62–67. Bibcode:2017CPL...690...62H. doi:10.1016/j.cplett.2017.10.034.
↑Jungmann R, Steinhauer C, Scheible M, Kuzyk A, Tinnefeld P, Simmel FC (November 2010). "Single-molecule kinetics and super-resolution microscopy by fluorescence imaging of transient binding on DNA origami". Nano Letters. 10 (11): 4756–61. Bibcode:2010NanoL..10.4756J. doi:10.1021/nl103427w. PMID20957983. S2CID11788360.
↑Prakash K (2021). "Laser-free super-resolution microscopy". Philosophical Transactions of the Royal Society A: Mathematical, Physical and Engineering Sciences. 379 (2199). bioRxiv10.1101/121061. doi:10.1098/rsta.2020.0144. PMID33896204.
↑Kaufmann R, Müller P, Hildenbrand G, Hausmann M, Cremer C (2010). "Analysis of Her2/neu membrane protein clusters in different types of breast cancer cells using localization microscopy". Journal of Microscopy. 242 (1): 46–54. doi:10.1111/j.1365-2818.2010.03436.x. PMID21118230. S2CID2119158.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.