Micrograph showing amyloid deposits (pink) in small bowel. Duodenum with amyloid deposition in lamina propria. Amyloid shows up as homogeneous pink material in lamina propria and around blood vessels. 20× magnification.
Amyloids are aggregates of proteins characterised by a fibrillar morphology of typically 7–13 nm in diameter, a β-sheetsecondary structure (known as cross-β) and ability to be stained by particular dyes, such as Congo red.[1] In the human body, amyloids have been linked to the development of various diseases.[2] Pathogenic amyloids form when previously healthy proteins lose their normal structure and physiological functions (misfolding) and form fibrous deposits within and around cells. These protein misfolding and deposition processes disrupt the healthy function of tissues and organs.
Such amyloids have been associated with (but not necessarily as the cause of) more than 50[2][3] human diseases, known as amyloidosis, and may play a role in some neurodegenerative diseases.[2][4] Some of these diseases are mainly sporadic and only a few cases are familial. Others are only familial. Some result from medical treatment. Prions are an infectious form of amyloids that can act as a template to convert other non-infectious forms.[5] Amyloids may also have normal biological functions; for example, in the formation of fimbriae in some genera of bacteria, transmission of epigenetic traits in fungi, as well as pigment deposition and hormone release in humans.[6]
Amyloids have been known to arise from many different proteins.[2][7] These polypeptide chains generally form β-sheet structures that aggregate into long fibers; however, identical polypeptides can fold into multiple distinct amyloid conformations.[8] The diversity of the conformations may have led to different forms of the prion diseases.[6]
An unusual secondary structure named α sheet has been proposed as the toxic constituent of amyloid precursor proteins,[9] but this idea is not widely accepted at present.
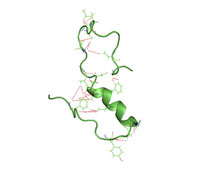
Amyloid of HET-s(218-289) prion pentamer, Podospora anserina (PDB: 2rnm)
Definition
The name amyloid comes from the early mistaken identification by Rudolf Virchow of the substance as starch (amylum in Latin, from Ancient Greek: ἄμυλον, romanized:amylon), based on crude iodine-staining techniques. For a period, the scientific community debated whether or not amyloid deposits are fatty deposits or carbohydrate deposits until it was finally found (in 1859) that they are, in fact, deposits of albumoid proteinaceous material.[10]
The classical, histopathological definition of amyloid is an extracellular, proteinaceous fibrillar deposit exhibiting β-sheetsecondary structure and identified by apple-green birefringence when stained with congo red under polarized light. These deposits often recruit various sugars and other components such as serum amyloid P component, resulting in complex, and sometimes inhomogeneous structures.[11] Recently this definition has come into question as some classic, amyloid species have been observed in distinctly intracellular locations.[12]
To date, 37 human proteins have been found to form amyloid in pathology and be associated with well-defined diseases.[2] The International Society of Amyloidosis classifies amyloid fibrils and their associated diseases based upon associated proteins (for example ATTR is the group of diseases and associated fibrils formed by TTR).[3] A table is included below.
Many examples of non-pathological amyloid with a well-defined physiological role have been identified in various organisms, including human. These may be termed as functional or physiological or native amyloid.[25][26][2]
Curlifibrils produced by E. coli,Salmonella, and a few other members of the Enterobacteriales (Csg). The genetic elements (operons) encoding the curli system are phylogenetic widespread and can be found in at least four bacterial phyla.[31] This suggest that many more bacteria may express curli fibrils.
GvpA, forming the walls of particular Gas vesicles, i.e. the buoyancy organelles of aquatic archaea and eubacteria[32]
Several yeast prions are based on an infectious amyloid, e.g. [PSI+] (Sup35p); [URE3] (Ure2p); [PIN+] or [RNQ+] (Rnq1p); [SWI1+] (Swi1p) and [OCT8+] (Cyc8p)
Structure of a fibril, consisting of one single protofilament, of the amyloid β peptide viewed down the long axis of the fibril (PDB: 2mlq)
Amyloids are formed of long unbranched fibers that are characterized by an extended β-sheet secondary structure in which individual β strands (β-strands) (coloured arrows in the adjacent figure) are arranged in an orientation perpendicular to the long axis of the fiber. Such a structure is known as cross-β structure. Each individual fiber may be 7–13 nanometres in width and a few micrometres in length.[6][2] The main hallmarks recognised by different disciplines to classify protein aggregates as amyloid is the presence of a fibrillar morphology with the expected diameter, detected using transmission electron microscopy (TEM) or atomic force microscopy (AFM), the presence of a cross-β secondary structure, determined with circular dichroism, FTIR, solid-state nuclear magnetic resonance (ssNMR), X-ray crystallography, or X-ray fiber diffraction (often considered the "gold-standard" test to see whether a structure contains cross-β fibres), and an ability to stain with specific dyes, such as Congo red, thioflavin T or thioflavin S.[2]
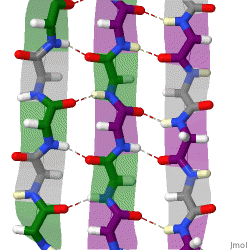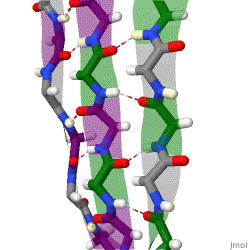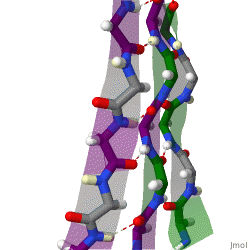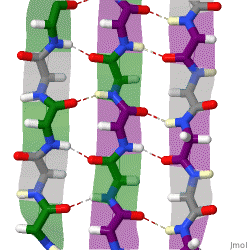
The term "cross-β" was based on the observation of two sets of diffraction lines, one longitudinal and one transverse, that form a characteristic "cross" pattern.[45] There are two characteristic scattering diffraction signals produced at 4.7 and 10 Å (0.47nm and 1.0nm), corresponding to the interstrand and stacking distances in β sheets.[1] The "stacks" of β sheet are short and traverse the breadth of the amyloid fibril; the length of the amyloid fibril is built by aligned β-strands. The cross-β pattern is considered a diagnostic hallmark of amyloid structure.[6]
Amyloid fibrils are generally composed of 1–8 protofilaments (one protofilament also corresponding to a fibril is shown in the figure), each 2–7nm in diameter, that interact laterally as flat ribbons that maintain the height of 2–7nm (that of a single protofilament) and are up to 30nm wide; more often protofilaments twist around each other to form the typically 7–13 nm wide fibrils.[2] Each protofilament possesses the typical cross-β structure and may be formed by 1–6 β-sheets (six are shown in the figure) stacked on each other. Each individual protein molecule can contribute one to several β-strands in each protofilament and the strands can be arranged in antiparallel β-sheets, but more often in parallel β-sheets. Only a fraction of the polypeptide chain is in a β-strand conformation in the fibrils, the remainder forms structured or unstructured loops or tails.
For a long time our knowledge of the atomic-level structure of amyloid fibrils was limited by the fact that they are unsuitable for the most traditional methods for studying protein structures. Recent years have seen progress in experimental methods, including solid-state NMR spectroscopy and Cryo-Electron Microscopy. Combined, these methods have provided 3D atomic structures of amyloid fibrils formed by amyloid β peptides, α-synuclein, tau, and the FUS protein, associated with various neurodegenerative diseases.[46][47]
X-ray diffraction studies of microcrystals revealed atomistic details of core region of amyloid, although only for simplified peptides having a length remarkably shorter than that of peptides or proteins involved in disease.[48][49] The crystallographic structures show that short stretches from amyloid-prone regions of amyloidogenic proteins run perpendicular to the filament axis, consistent with the "cross-β" feature of amyloid structure. They also reveal a number of characteristics of amyloid structures – neighboring β-sheets are tightly packed together via an interface devoid of water (therefore referred to as dry interface), with the opposing β-strands slightly offset from each other such that their side-chains interdigitate. This compact dehydrated interface created was termed a steric-zipper interface.[6] There are eight theoretical classes of steric-zipper interfaces, dictated by the directionality of the β-sheets (parallel and anti-parallel) and symmetry between adjacent β-sheets. A limitation of X-ray crystallography for solving amyloid structure is represented by the need to form microcrystals, which can be achieved only with peptides shorter than those associated with disease.
Although bona fide amyloid structures always are based on intermolecular β-sheets, different types of "higher order" tertiary folds have been observed or proposed. The β-sheets may form a β-sandwich, or a β-solenoid which may be either β-helix or β-roll. Native-like amyloid fibrils in which native β-sheet containing proteins maintain their native-like structure in the fibrils have also been proposed.[50] There are few developed ideas on how the complex backbone topologies of disulfide-constrained proteins, which are prone to form amyloid fibrils (such as insulin and lysozyme), adopt the amyloid β-sheet motif. The presence of multiple constraints significantly reduces the accessible conformational space, making computational simulations of amyloid structures more feasible. [51]
One complicating factor in studies of amyloidogenic polypeptides is that identical polypeptides can fold into multiple distinct amyloid conformations.[6] This phenomenon is typically described as amyloid polymorphism.[8][52][53] It has notable biological consequences given that it is thought to explain the prion strain phenomenon.
Amyloid is formed through the polymerization of hundreds to thousands of monomeric peptides or proteins into long fibers. Amyloid formation involves a lag phase (also called nucleation phase), an exponential phase (also called growth phase) and a plateau phase (also called saturation phase), as shown in the figure.[54][55][56][57] Indeed, when the quantity of fibrils is plotted versus time, a sigmoidal time course is observed reflecting the three distinct phases.
In the simplest model of 'nucleated polymerization' (marked by red arrows in the figure below), individual unfolded or partially unfolded polypeptide chains (monomers) convert into a nucleus (monomer or oligomer) via a thermodynamically unfavourable process that occurs early in the lag phase.[56] Fibrils grow subsequently from these nuclei through the addition of monomers in the exponential phase.[56]
A different model, called 'nucleated conformational conversion' and marked by blue arrows in the figure below, was introduced later on to fit some experimental observations: monomers have often been found to convert rapidly into misfolded and highly disorganized oligomers distinct from nuclei.[58] Only later on, will these aggregates reorganise structurally into nuclei, on which other disorganised oligomers will add and reorganise through a templating or induced-fit mechanism (this 'nucleated conformational conversion' model), eventually forming fibrils.[58]
Normally folded proteins have to unfold partially before aggregation can take place through one of these mechanisms.[59] In some cases, however, folded proteins can aggregate without crossing the major energy barrier for unfolding, by populating native-like conformations as a consequence of thermal fluctuations, ligand release or local unfolding occurring in particular circumstances.[59] In these native-like conformations, segments that are normally buried or structured in the fully folded and possessing a high propensity to aggregate become exposed to the solvent or flexible, allowing the formation of native-like aggregates, which convert subsequently into nuclei and fibrils. This process is called 'native-like aggregation' (green arrows in the figure) and is similar to the 'nucleated conformational conversion' model.
A more recent, modern and thorough model of amyloid fibril formation involves the intervention of secondary events, such as 'fragmentation', in which a fibril breaks into two or more shorter fibrils, and 'secondary nucleation', in which fibril surfaces (not fibril ends) catalyze the formation of new nuclei.[57] Both secondary events increase the number of fibril ends able to recruit new monomers or oligomers, therefore accelerating fibril formation through a positive feedback mechanism. These events add to the well recognised steps of primary nucleation (formation of the nucleus from the monomers through one of models described above), fibril elongation (addition of monomers or oligomers to growing fibril ends) and dissociation (opposite process).
Such a new model is described in the figure on the right and involves the utilization of a master equation that includes all steps of amyloid fibril formation, i.e. primary nucleation, fibril elongation, secondary nucleation and fibril fragmentation.[57][60] The rate constants of the various steps can be determined from a global fit of a number of time courses of aggregation (for example ThT fluorescence emission versus time) recorded at different protein concentrations.[57] The general master equation approach to amyloid fibril formation with secondary pathways has been developed by Knowles, Vendruscolo, Cohen, Michaels and coworkers and considers the time evolution of the concentration of fibrils of length (here represents the number of monomers in an aggregate).[60]where denotes the Kronecker delta. The physical interpretation of the various terms in the above master equation is straight forward: the terms on the first line describe the growth of fibrils via monomer addition with rate constant (elongation). The terms on the second line describe monomer dissociation, i.e. the inverse process of elongation. is the rate constant of monomer dissociation. The terms on the third line describe the effect of fragmentation, which is assumed to occur homogeneously along fibrils with rate constant . Finally, the terms on the last line describe primary and secondary nucleation respectively. Note that the rate of secondary nucleation is proportional to the mass of aggregates, defined as .
Following this analytical approach, it has become apparent that the lag phase does not correspond necessarily to only nucleus formation, but rather results from a combination of various steps. Similarly, the exponential phase is not only fibril elongation, but results from a combination of various steps, involving primary nucleation, fibril elongation, but also secondary events. A significant quantity of fibrils resulting from primary nucleation and fibril elongation may be formed during the lag phase and secondary steps, rather than only fibril elongation, can be the dominant processes contributing to fibril growth during the exponential phase. With this new model, any perturbing agents of amyloid fibril formation, such as putative drugs, metabolites, mutations, chaperones, etc., can be assigned to a specific step of fibril formation.
Amino acid sequence and amyloid formation
In general, amyloid polymerization (aggregation or non-covalent polymerization) is sequence-sensitive, that is mutations in the sequence can induce or prevent self-assembly.[61][62] For example, humans produce amylin, an amyloidogenic peptide associated with type II diabetes, but in rats and mice prolines are substituted in critical locations and amyloidogenesis does not occur.[63] Studies comparing synthetic to recombinant β amyloid peptide in assays measuring rate of fibrillation, fibril homogeneity, and cellular toxicity showed that recombinant β amyloid peptide has a faster fibrillation rate and greater toxicity than synthetic β amyloid peptide.[64]
There are multiple classes of amyloid-forming polypeptide sequences.[8][52][53] Glutamine-rich polypeptides are important in the amyloidogenesis of Yeast and mammalian prions, as well as trinucleotide repeat disorders including Huntington's disease. When glutamine-rich polypeptides are in a β-sheet conformation, glutamines can brace the structure by forming inter-strand hydrogen bonding between its amide carbonyls and nitrogens of both the backbone and side chains. The onset age for Huntington's disease shows an inverse correlation with the length of the polyglutamine sequence, with analogous findings in a C. elegans model system with engineered polyglutamine peptides.[65]
Other polypeptides and proteins such as amylin and the β amyloid peptide do not have a simple consensus sequence and are thought to aggregate through the sequence segments enriched with hydrophobic residues, or residues with high propensity to form β-sheet structure.[61] Among the hydrophobic residues, aromatic amino-acids are found to have the highest amyloidogenic propensity.[66][67]
Cross-polymerization (fibrils of one polypeptide sequence causing other fibrils of another sequence to form) is observed in vitro and possibly in vivo. This phenomenon is important, since it would explain interspecies prion propagation and differential rates of prion propagation, as well as a statistical link between Alzheimer's and type 2 diabetes.[68] In general, the more similar the peptide sequence the more efficient cross-polymerization is, though entirely dissimilar sequences can cross-polymerize and highly similar sequences can even be "blockers" that prevent polymerization.[citation needed]
Amyloid toxicity
The reasons why amyloid cause diseases are unclear. In some cases, the deposits physically disrupt tissue architecture, suggesting disruption of function by some bulk process. An emerging consensus implicates prefibrillar intermediates, rather than mature amyloid fibers, in causing cell death, particularly in neurodegenerative diseases.[17][69] The fibrils are, however, far from innocuous, as they keep the protein homeostasis network engaged, release oligomers, cause the formation of toxic oligomers via secondary nucleation, grow indefinitely spreading from district to district[2] and, in some cases, may be toxic themselves.[70]
Calcium dysregulation has been observed to occur early in cells exposed to protein oligomers. These small aggregates can form ion channels through lipid bilayer membranes and activate NMDA and AMPA receptors. Channel formation has been hypothesized to account for calcium dysregulation and mitochondrial dysfunction by allowing indiscriminate leakage of ions across cell membranes.[71] Studies have shown that amyloid deposition is associated with mitochondrial dysfunction and a resulting generation of reactive oxygen species (ROS), which can initiate a signalling pathway leading to apoptosis.[72] There are reports that indicate amyloid polymers (such as those of huntingtin, associated with Huntington's disease) can induce the polymerization of essential amyloidogenic proteins, which should be deleterious to cells. Also, interaction partners of these essential proteins can also be sequestered.[73]
All these mechanisms of toxicity are likely to play a role. In fact, the aggregation of a protein generates a variety of aggregates, all of which are likely to be toxic to some degree. A wide variety of biochemical, physiological and cytological perturbations has been identified following the exposure of cells and animals to such species, independently of their identity. The oligomers have also been reported to interact with a variety of molecular targets. Hence, it is unlikely that there is a unique mechanism of toxicity or a unique cascade of cellular events. The misfolded nature of protein aggregates causes a multitude of aberrant interactions with a multitude of cellular components, including membranes, protein receptors, soluble proteins, RNAs, small metabolites, etc.
Histological staining
In the clinical setting, amyloid diseases are typically identified by a change in the spectroscopic properties of planar aromaticdyes such as thioflavin T, congo red or NIAD-4.[74] In general, this is attributed to the environmental change, as these dyes intercalate between β-strands to confine their structure.[75]
Congo Red positivity remains the gold standard for diagnosis of amyloidosis. In general, binding of Congo Red to amyloid plaques produces a typical apple-green birefringence when viewed under cross-polarized light. Recently, significant enhancement of fluorescence quantum yield of NIAD-4 was exploited to super-resolution fluorescence imaging of amyloid fibrils[76] and oligomers.[77] To avoid nonspecific staining, other histology stains, such as the hematoxylin and eosin stain, are used to quench the dyes' activity in other places such as the nucleus, where the dye might bind. Modern antibody technology and immunohistochemistry has made specific staining easier, but often this can cause trouble because epitopes can be concealed in the amyloid fold; in general, an amyloid protein structure is a different conformation from the one that the antibody recognizes.
The beta sheet is a common motif of the regular protein secondary structure. Beta sheets consist of beta strands (β-strands) connected laterally by at least two or three backbone hydrogen bonds, forming a generally twisted, pleated sheet. A β-strand is a stretch of polypeptide chain typically 3 to 10 amino acids long with backbone in an extended conformation. The supramolecular association of β-sheets has been implicated in the formation of the fibrils and protein aggregates observed in amyloidosis, Alzheimer's disease and other proteinopathies.
A prion is a misfolded protein that induces misfolding in normal variants of the same protein, leading to cellular death. Prions are responsible for prion diseases, known as transmissible spongiform encephalopathy (TSEs), which are fatal and transmissible neurodegenerative diseases affecting both humans and animals. These proteins can misfold sporadically, due to genetic mutations, or by exposure to an already misfolded protein, leading to an abnormal three-dimensional structure that can propagate misfolding in other proteins.
Protein folding is the physical process by which a protein, after synthesis by a ribosome as a linear chain of amino acids, changes from an unstable random coil into a more ordered three-dimensional structure. This structure permits the protein to become biologically functional.
Alpha-synuclein (aSyn) is a protein that, in humans, is encoded by the SNCA gene. Alpha-synuclein is a neuronal protein that regulates synaptic vesicle trafficking and subsequent neurotransmitter release.
Transthyretin (TTR or TBPA) is a transport protein in the plasma and cerebrospinal fluid that transports the thyroid hormone thyroxine (T4) and retinol to the liver. This is how transthyretin gained its name: transports thyroxine and retinol. The liver secretes TTR into the blood, and the choroid plexus secretes TTR into the cerebrospinal fluid.
Amylin, or islet amyloid polypeptide (IAPP), is a 37-residue peptide hormone. It is co-secreted with insulin from the pancreatic β-cells in the ratio of approximately 100:1 (insulin:amylin). Amylin plays a role in glycemic regulation by slowing gastric emptying and promoting satiety, thereby preventing post-prandial spikes in blood glucose levels.
Amyloid beta denotes peptides of 36–43 amino acids that are the main component of the amyloid plaques found in the brains of people with Alzheimer's disease. The peptides derive from the amyloid-beta precursor protein (APP), which is cleaved by beta secretase and gamma secretase to yield Aβ in a cholesterol-dependent process and substrate presentation. Both neurons and oligodendrocytes produce and release Aβ in the brain, contributing to formation of amyloid plaques. Aβ molecules can aggregate to form flexible soluble oligomers which may exist in several forms. It is now believed that certain misfolded oligomers can induce other Aβ molecules to also take the misfolded oligomeric form, leading to a chain reaction akin to a prion infection. The oligomers are toxic to nerve cells. The other protein implicated in Alzheimer's disease, tau protein, also forms such prion-like misfolded oligomers, and there is some evidence that misfolded Aβ can induce tau to misfold.
The major prion protein (PrP) is encoded in the human body by the PRNP gene also known as CD230. Expression of the protein is most predominant in the nervous system but occurs in many other tissues throughout the body.
Thioflavins are fluorescent dyes that are available as at least two compounds, namely Thioflavin T and Thioflavin S. Both are used for histology staining and biophysical studies of protein aggregation. In particular, these dyes have been used since 1989 to investigate amyloid formation. They are also used in biophysical studies of the electrophysiology of bacteria. Thioflavins are corrosive, irritant, and acutely toxic, causing serious eye damage. Thioflavin T has been used in research into Alzheimer's disease and other neurodegenerative diseases.
A fungal prion is a prion that infects hosts which are fungi. Fungal prions are naturally occurring proteins that can switch between multiple, structurally distinct conformations, at least one of which is self-propagating and transmissible to other prions. This transmission of protein state represents an epigenetic phenomenon where information is encoded in the protein structure itself, instead of in nucleic acids. Several prion-forming proteins have been identified in fungi, primarily in the yeast Saccharomyces cerevisiae. These fungal prions are generally considered benign, and in some cases even confer a selectable advantage to the organism.
Physalaemin is a tachykinin peptide obtained from the Physalaemus frog, closely related to substance P. Its structure was first elucidated in 1964.
The biochemistry of Alzheimer's disease, the most common cause of dementia, is not yet very well understood. Alzheimer's disease (AD) has been identified as a proteopathy: a protein misfolding disease due to the accumulation of abnormally folded amyloid beta (Aβ) protein in the brain. Amyloid beta is a short peptide that is an abnormal proteolytic byproduct of the transmembrane protein amyloid-beta precursor protein (APP), whose function is unclear but thought to be involved in neuronal development. The presenilins are components of proteolytic complex involved in APP processing and degradation.
Alpha sheet is an atypical secondary structure in proteins, first proposed by Linus Pauling and Robert Corey in 1951. The hydrogen bonding pattern in an alpha sheet is similar to that of a beta sheet, but the orientation of the carbonyl and amino groups in the peptide bond units is distinctive; in a single strand, all the carbonyl groups are oriented in the same direction on one side of the pleat, and all the amino groups are oriented in the same direction on the opposite side of the sheet. Thus the alpha sheet accumulates an inherent separation of electrostatic charge, with one edge of the sheet exposing negatively charged carbonyl groups and the opposite edge exposing positively charged amino groups. Unlike the alpha helix and beta sheet, the alpha sheet configuration does not require all component amino acid residues to lie within a single region of dihedral angles; instead, the alpha sheet contains residues of alternating dihedrals in the traditional right-handed (αR) and left-handed (αL) helical regions of Ramachandran space. Although the alpha sheet is only rarely observed in natural protein structures, it has been speculated to play a role in amyloid disease and it was found to be a stable form for amyloidogenic proteins in molecular dynamics simulations. Alpha sheets have also been observed in X-ray crystallography structures of designed peptides.
In medicine, proteinopathy, or proteopathy, protein conformational disorder, or protein misfolding disease, is a class of diseases in which certain proteins become structurally abnormal, and thereby disrupt the function of cells, tissues and organs of the body.
In molecular biology, protein aggregation is a phenomenon in which intrinsically-disordered or mis-folded proteins aggregate either intra- or extracellularly. Protein aggregates have been implicated in a wide variety of diseases known as amyloidoses, including ALS, Alzheimer's, Parkinson's and prion disease.
p3 peptide also known as amyloid β- peptide (Aβ)17–40/42 is the peptide resulting from the α- and γ-secretase cleavage from the amyloid precursor protein (APP). It is known to be the major constituent of diffuse plaques observed in Alzheimer's disease (AD) brains and pre-amyloid plaques in people affected by Down syndrome. However, p3 peptide's role in these diseases is not truly known yet.
The ion channel hypothesis of Alzheimer's disease (AD), also known as the channel hypothesis or the amyloid beta ion channel hypothesis, is a more recent variant of the amyloid hypothesis of AD, which identifies amyloid beta (Aβ) as the underlying cause of neurotoxicity seen in AD. While the traditional formulation of the amyloid hypothesis pinpoints insoluble, fibrillar aggregates of Aβ as the basis of disruption of calcium ion homeostasis and subsequent apoptosis in AD, the ion channel hypothesis in 1993 introduced the possibility of an ion-channel-forming oligomer of soluble, non-fibrillar Aβ as the cytotoxic species allowing unregulated calcium influx into neurons in AD.
Hsp104 is a heat-shock protein. It is known to reverse toxicity of mutant α-synuclein, TDP-43, FUS, and TAF15 in yeast cells. Conserved in prokaryotes (ClpB), fungi, plants and as well as animal mitochondria, there is yet to see hsp104 in multicellular animals. Hsp104 is classified as a. AAA+ ATPases and a subgroup of Hsp100/Clp, because of the usage of Atp hydrolysis for structural modulation of other proteins. Hsp104 is not needed for normal cell growth but when exposed to stress there is an increase amount. Removing the aggregates without the hsp104 is insufficient there highlighting the importance of this heat shock protein and its interactions.
The Curli protein is a type of amyloid fiber produced by certain strains of enterobacteria. They are extracellular fibers located on bacteria such as E. coli and Salmonella spp. These fibers serve to promote cell community behavior through biofilm formation in the extracellular matrix. Amyloids are associated with several human neurodegenerative diseases such as Alzheimer's disease, Huntington's disease, Parkinson's disease, and prion diseases. The study of curli may help to understand human diseases thought to arise from improper amyloid fiber formation. The curli pili are generally assembled through the extracellular nucleation precipitation pathway.
Computational methods that use protein sequence and/ or protein structure to predict protein aggregation. The table below, shows the main features of software for prediction of protein aggregation
References
1 2 Sunde M, Serpell LC, Bartlam M, Fraser PE, Pepys MB, Blake CC (October 1997). "Common core structure of amyloid fibrils by synchrotron X-ray diffraction". Journal of Molecular Biology. 273 (3): 729–39. doi:10.1006/jmbi.1997.1348. PMID9356260. S2CID19394482.
1 2 3 Balbach JJ, Ishii Y, Antzutkin ON, Leapman RD, Rizzo NW, Dyda F, etal. (November 2000). "Amyloid fibril formation by Aβ16-22, a seven-residue fragment of the Alzheimer's β-amyloid peptide, and structural characterization by solid state NMR". Biochemistry. 39 (45): 13748–59. doi:10.1021/bi0011330. PMID11076514. S2CID17232045.
↑ Chiang PK, Lam MA, Luo Y (September 2008). "The many faces of amyloid β in Alzheimer's disease". Current Molecular Medicine. 8 (6): 580–4. doi:10.2174/156652408785747951. PMID18781964.
↑ Weydt P, La Spada AR (August 2006). "Targeting protein aggregation in neurodegeneration--lessons from polyglutamine disorders". Expert Opinion on Therapeutic Targets. 10 (4): 505–13. doi:10.1517/14728222.10.4.505. PMID16848688. S2CID24483289.
↑ Höppener JW, Ahrén B, Lips CJ (August 2000). "Islet amyloid and type 2 diabetes mellitus". The New England Journal of Medicine. 343 (6): 411–9. doi:10.1056/NEJM200008103430607. PMID10933741.
↑ Serag AA, Altenbach C, Gingery M, Hubbell WL, Yeates TO (October 2002). "Arrangement of subunits and ordering of β-strands in an amyloid sheet". Nature Structural Biology. 9 (10): 734–9. doi:10.1038/nsb838. PMID12219081. S2CID23926428.
↑ Jarrett JT, Berger EP, Lansbury PT (May 1993). "The carboxy terminus of the β amyloid protein is critical for the seeding of amyloid formation: implications for the pathogenesis of Alzheimer's disease". Biochemistry. 32 (18): 4693–7. doi:10.1021/bi00069a001. PMID8490014.
↑ Finder VH, Vodopivec I, Nitsch RM, Glockshuber R (February 2010). "The recombinant amyloid-β peptide Aβ1-42 aggregates faster and is more neurotoxic than synthetic Aβ-42". Journal of Molecular Biology. 396 (1): 9–18. doi:10.1016/j.jmb.2009.12.016. PMID20026079.
↑ Pawar AP, Dubay KF, Zurdo J, Chiti F, Vendruscolo M, Dobson CM (July 2005). "Prediction of "aggregation-prone" and "aggregation-susceptible" regions in proteins associated with neurodegenerative diseases". Journal of Molecular Biology. 350 (2): 379–92. doi:10.1016/j.jmb.2005.04.016. PMID15925383.
↑ Huh H, Lee J, Kim HJ, Hohng S, Kim SK (2017). "Morphological analysis of oligomeric vs. fibrillar forms of α-synuclein aggregates with super-resolution BALM imaging". Chemical Physics Letters. 690: 62–67. Bibcode:2017CPL...690...62H. doi:10.1016/j.cplett.2017.10.034.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.