
Fibrinogen is a glycoprotein complex, produced in the liver, that circulates in the blood of all vertebrates. During tissue and vascular injury, it is converted enzymatically by thrombin to fibrin and then to a fibrin-based blood clot. Fibrin clots function primarily to occlude blood vessels to stop bleeding. Fibrin also binds and reduces the activity of thrombin. This activity, sometimes referred to as antithrombin I, limits clotting. Fibrin also mediates blood platelet and endothelial cell spreading, tissue fibroblast proliferation, capillary tube formation, and angiogenesis and thereby promotes revascularization and wound healing.
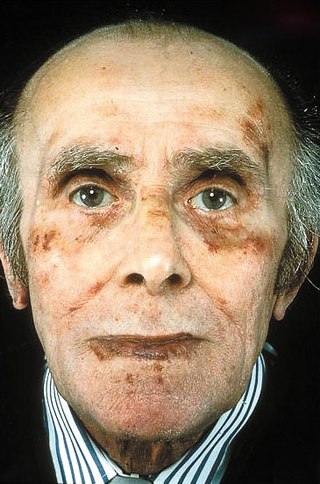
Amyloidosis is a group of diseases in which abnormal proteins, known as amyloid fibrils, build up in tissue. There are several non-specific and vague signs and symptoms associated with amyloidosis. These include fatigue, peripheral edema, weight loss, shortness of breath, palpitations, and feeling faint with standing. In AL amyloidosis, specific indicators can include enlargement of the tongue and periorbital purpura. In wild-type ATTR amyloidosis, non-cardiac symptoms include: bilateral carpal tunnel syndrome, lumbar spinal stenosis, biceps tendon rupture, small fiber neuropathy, and autonomic dysfunction.

Alport syndrome is a genetic disorder affecting around 1 in 5,000-10,000 children, characterized by glomerulonephritis, end-stage kidney disease, and hearing loss. Alport syndrome can also affect the eyes, though the changes do not usually affect vision, except when changes to the lens occur in later life. Blood in urine is universal. Proteinuria is a feature as kidney disease progresses.

Transthyretin (TTR or TBPA) is a transport protein in the plasma and cerebrospinal fluid that transports the thyroid hormone thyroxine (T4) and retinol to the liver. This is how transthyretin gained its name: transports thyroxine and retinol. The liver secretes TTR into the blood, and the choroid plexus secretes TTR into the cerebrospinal fluid.

Renal tubular acidosis (RTA) is a medical condition that involves an accumulation of acid in the body due to a failure of the kidneys to appropriately acidify the urine. In renal physiology, when blood is filtered by the kidney, the filtrate passes through the tubules of the nephron, allowing for exchange of salts, acid equivalents, and other solutes before it drains into the bladder as urine. The metabolic acidosis that results from RTA may be caused either by insufficient secretion of hydrogen ions into the latter portions of the nephron or by failure to reabsorb sufficient bicarbonate ions from the filtrate in the early portion of the nephron. Although a metabolic acidosis also occurs in those with chronic kidney disease, the term RTA is reserved for individuals with poor urinary acidification in otherwise well-functioning kidneys. Several different types of RTA exist, which all have different syndromes and different causes. RTA is usually an incidental finding based on routine blood draws that show abnormal results. Clinically, patients may present with vague symptoms such as dehydration, mental status changes, or delayed growth in adolescents.

Familial amyloid polyneuropathy, also called transthyretin-related hereditary amyloidosis, transthyretin amyloidosis abbreviated also as ATTR, or Corino de Andrade's disease, is an autosomal dominant neurodegenerative disease. It is a form of amyloidosis, and was first identified and described by Portuguese neurologist Mário Corino da Costa Andrade, in 1952. FAP is distinct from senile systemic amyloidosis (SSA), which is not inherited, and which was determined to be the primary cause of death for 70% of supercentenarians who have been autopsied. FAP can be ameliorated by liver transplantation.

Cardiac amyloidosis is a subcategory of amyloidosis where there is depositing of the protein amyloid in the cardiac muscle and surrounding tissues. Amyloid, a misfolded and insoluble protein, can become a deposit in the heart's atria, valves, or ventricles. These deposits can cause thickening of different sections of the heart, leading to decreased cardiac function. The overall decrease in cardiac function leads to a plethora of symptoms. This multisystem disease was often misdiagnosed, with a corrected analysis only during autopsy. Advancements of technologies have increased earlier accuracy of diagnosis. Cardiac amyloidosis has multiple sub-types including light chain, familial, and senile. One of the most studied types is light chain cardiac amyloidosis. Prognosis depends on the extent of the deposits in the body and the type of amyloidosis. New treatment methods are actively being researched in regards to the treatment of heart failure and specific cardiac amyloidosis problems.

Alpha sheet is an atypical secondary structure in proteins, first proposed by Linus Pauling and Robert Corey in 1951. The hydrogen bonding pattern in an alpha sheet is similar to that of a beta sheet, but the orientation of the carbonyl and amino groups in the peptide bond units is distinctive; in a single strand, all the carbonyl groups are oriented in the same direction on one side of the pleat, and all the amino groups are oriented in the same direction on the opposite side of the sheet. Thus the alpha sheet accumulates an inherent separation of electrostatic charge, with one edge of the sheet exposing negatively charged carbonyl groups and the opposite edge exposing positively charged amino groups. Unlike the alpha helix and beta sheet, the alpha sheet configuration does not require all component amino acid residues to lie within a single region of dihedral angles; instead, the alpha sheet contains residues of alternating dihedrals in the traditional right-handed (αR) and left-handed (αL) helical regions of Ramachandran space. Although the alpha sheet is only rarely observed in natural protein structures, it has been speculated to play a role in amyloid disease and it was found to be a stable form for amyloidogenic proteins in molecular dynamics simulations. Alpha sheets have also been observed in X-ray crystallography structures of designed peptides.
The dysfibrinogenemias consist of three types of fibrinogen disorders in which a critical blood clotting factor, fibrinogen, circulates at normal levels but is dysfunctional. Congenital dysfibrinogenemia is an inherited disorder in which one of the parental genes produces an abnormal fibrinogen. This fibrinogen interferes with normal blood clotting and/or lysis of blood clots. The condition therefore may cause pathological bleeding and/or thrombosis. Acquired dysfibrinogenemia is a non-hereditary disorder in which fibrinogen is dysfunctional due to the presence of liver disease, autoimmune disease, a plasma cell dyscrasias, or certain cancers. It is associated primarily with pathological bleeding. Hereditary fibrinogen Aα-Chain amyloidosis is a sub-category of congenital dysfibrinogenemia in which the dysfunctional fibrinogen does not cause bleeding or thrombosis but rather gradually accumulates in, and disrupts the function of, the kidney.

Fibrinogen alpha chain is a protein that in humans is encoded by the FGA gene.

Leukocyte cell-derived chemotaxin-2 (LECT2) is a protein first described in 1996 as a chemotactic factor for neutrophils, i.e. it stimulated human neutrophils to move directionally in an in vitro assay system. The protein was detected in and purified from cultures of Phytohaemagglutinin-activated human T-cell leukemia SKW-3 cells. Subsequent studies have defined LECT2 as a hepatokine, i.e. a substance made and released into the circulation by liver hepatocyte cells that regulates the function of other cells: it is a hepatocyte-derived, hormone-like, signaling protein.
In hematology, plasma cell dyscrasias are a spectrum of progressively more severe monoclonal gammopathies in which a clone or multiple clones of pre-malignant or malignant plasma cells over-produce and secrete into the blood stream a myeloma protein, i.e. an abnormal monoclonal antibody or portion thereof. The exception to this rule is the disorder termed non-secretory multiple myeloma; this disorder is a form of plasma cell dyscrasia in which no myeloma protein is detected in serum or urine of individuals who have clear evidence of an increase in clonal bone marrow plasma cells and/or evidence of clonal plasma cell-mediated tissue injury. Here, a clone of plasma cells refers to group of plasma cells that are abnormal in that they have an identical genetic identity and therefore are descendants of a single genetically distinct ancestor cell.
Amyloid light-chain (AL) amyloidosis, also known as primary amyloidosis, is the most common form of systemic amyloidosis. The disease is caused when a person's antibody-producing cells do not function properly and produce abnormal protein fibers made of components of antibodies called light chains. These light chains come together to form amyloid deposits which can cause serious damage to different organs. An abnormal light chain in urine is known as Bence Jones protein.
AA amyloidosis is a form of amyloidosis, a disease characterized by the abnormal deposition of fibers of insoluble protein in the extracellular space of various tissues and organs. In AA amyloidosis, the deposited protein is serum amyloid A protein (SAA), an acute-phase protein which is normally soluble and whose plasma concentration is highest during inflammation.
The familial amyloid neuropathies are a rare group of autosomal dominant diseases wherein the autonomic nervous system and/or other nerves are compromised by protein aggregation and/or amyloid fibril formation.
Sir Mark Brian Pepys is a South African-born British academic of medicine. He was until 2011 Professor of Medicine at University College London and Head of Medicine at the Hampstead Campus and the Royal Free Hospital.
Fanconi syndrome or Fanconi's syndrome is a syndrome of inadequate reabsorption in the proximal renal tubules of the kidney. The syndrome can be caused by various underlying congenital or acquired diseases, by toxicity, or by adverse drug reactions. It results in various small molecules of metabolism being passed into the urine instead of being reabsorbed from the tubular fluid. Fanconi syndrome affects the proximal tubules, namely, the proximal convoluted tubule (PCT), which is the first part of the tubule to process fluid after it is filtered through the glomerulus, and the proximal straight tubule, which leads to the descending limb of loop of Henle.

Light chain deposition disease (LCDD) is a rare blood cell disease which is characterized by deposition of fragments of infection-fighting immunoglobulins, called light chains (LCs), in the body. LCs are normally cleared by the kidneys, but in LCDD, these light chain deposits damage organs and cause disease. The kidneys are almost always affected and this often leads to kidney failure. About half of people with light chain deposition disease also have a plasma cell dyscrasia, a spectrum of diseases that includes multiple myeloma, Waldenström's macroglobulinemia, and the monoclonal gammopathy of undetermined significance premalignant stages of these two diseases. Unlike in AL amyloidosis, in which light chains are laid down in characteristic amyloid deposits, in LCDD, light chains are deposited in non-amyloid granules.

LECT2 Amyloidosis (ALECT2) is a form of amyloidosis caused by the LECT2 protein. It was found to be the third most common cause of amyloidosis in a set of more than 4,000 individuals studied at the Mayo Clinic; the first and second most common forms the disorder were AL amyloidosis and AA amyloidosis, respectively. Amyloidosis is a disorder in which the abnormal deposition of a protein in organs and/or tissues gradually leads to organ failure and/or tissue injury.
