
IgA nephropathy (IgAN), also known as Berger's disease, or synpharyngitic glomerulonephritis, is a disease of the kidney and the immune system; specifically it is a form of glomerulonephritis or an inflammation of the glomeruli of the kidney. Aggressive Berger's disease can attack other major organs, such as the liver, skin and heart.

The alternative pathway is a type of cascade reaction of the complement system and is a component of the innate immune system, a natural defense against infections.
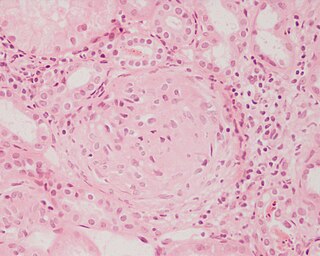
Glomerulonephritis (GN) is a term used to refer to several kidney diseases. Many of the diseases are characterised by inflammation either of the glomeruli or of the small blood vessels in the kidneys, hence the name, but not all diseases necessarily have an inflammatory component.

Membranous glomerulonephritis (MGN) is a slowly progressive disease of the kidney affecting mostly people between ages of 30 and 50 years, usually white people.

Nephritic syndrome is a syndrome comprising signs of nephritis, which is kidney disease involving inflammation. It often occurs in the glomerulus, where it is called glomerulonephritis. Glomerulonephritis is characterized by inflammation and thinning of the glomerular basement membrane and the occurrence of small pores in the podocytes of the glomerulus. These pores become large enough to permit both proteins and red blood cells to pass into the urine. By contrast, nephrotic syndrome is characterized by proteinuria and a constellation of other symptoms that specifically do not include hematuria. Nephritic syndrome, like nephrotic syndrome, may involve low level of albumin in the blood due to the protein albumin moving from the blood to the urine.

Focal segmental glomerulosclerosis (FSGS) is a histopathologic finding of scarring (sclerosis) of glomeruli and damage to renal podocytes. This process damages the filtration function of the kidney, resulting in protein presence in the urine due to protein loss. FSGS is a leading cause of excess protein loss—nephrotic syndrome—in children and adults. Signs and symptoms include proteinuria and edema. Kidney failure is a common long-term complication of the disease. FSGS can be classified as primary, secondary, or genetic, depending on whether a particular toxic or pathologic stressor or genetic predisposition can be identified as the cause. Diagnosis is established by renal biopsy, and treatment consists of glucocorticoids and other immune-modulatory drugs. Response to therapy is variable, with a significant portion of patients progressing to end-stage kidney failure. FSGS is estimated to occur in 7 persons per million, with males and African peoples at higher risk.

Complement component 3, often simply called C3, is a protein of the immune system that is found primarily in the blood. It plays a central role in the complement system of vertebrate animals and contributes to innate immunity. In humans it is encoded on chromosome 19 by a gene called C3.

Acute proliferative glomerulonephritis is a disorder of the small blood vessels of the kidney. It is a common complication of bacterial infections, typically skin infection by Streptococcus bacteria types 12, 4 and 1 (impetigo) but also after streptococcal pharyngitis, for which it is also known as postinfectious glomerulonephritis (PIGN) or poststreptococcal glomerulonephritis (PSGN). It can be a risk factor for future albuminuria. In adults, the signs and symptoms of infection may still be present at the time when the kidney problems develop, and the terms infection-related glomerulonephritis or bacterial infection-related glomerulonephritis are also used. Acute glomerulonephritis resulted in 19,000 deaths in 2013, down from 24,000 deaths in 1990 worldwide.
C3b is the larger of two elements formed by the cleavage of complement component 3, and is considered an important part of the innate immune system. C3b is potent in opsonization: tagging pathogens, immune complexes (antigen-antibody), and apoptotic cells for phagocytosis. Additionally, C3b plays a role in forming a C3 convertase when bound to Factor B, or a C5 convertase when bound to C4b and C2b or when an additional C3b molecule binds to the C3bBb complex.
Barraquer–Simons syndrome is a rare form of lipodystrophy, which usually first affects the head, and then spreads to the thorax. It is named for Luis Barraquer Roviralta (1855–1928), a Spanish physician, and Arthur Simons (1879–1942), a German physician. Some evidence links it to LMNB2.

Complement factor H-related protein 5 is a protein that in humans is encoded by the CFHR5 gene.

Type III hypersensitivity, in the Gell and Coombs classification of allergic reactions, occurs when there is accumulation of immune complexes that have not been adequately cleared by innate immune cells, giving rise to an inflammatory response and attraction of leukocytes. There are three steps that lead to this response. The first step is immune complex formation, which involves the binding of antigens to antibodies to form mobile immune complexes. The second step is immune complex deposition, during which the complexes leave the plasma and are deposited into tissues. Finally, the third step is the inflammatory reaction, during which the classical pathway is activated and macrophages and neutrophils are recruited to the affected tissues. Such reactions may progress to immune complex diseases.

Mesangial proliferative glomerulonephritis (MesPGN) is a morphological pattern characterized by a numerical increase in mesangial cells and expansion of the extracellular matrix within the mesangium of the glomerulus. The increase in the number of mesangial cells can be diffuse or local and immunoglobulin and/or complement deposition can also occur. MesPGN is associated with a variety of disease processes affecting the glomerulus, though can be idiopathic. The clinical presentation of MesPGN usually consists of hematuria or nephrotic syndrome. Treatment is often consistent with the histologic pattern of and/or disease process contributing to mesangial proliferative glomerulonephritis, and usually involves some form of immunosuppressant.
Diffuse proliferative glomerulonephritis (DPGN) is a type of glomerulonephritis that is the most serious form of renal lesions in SLE and is also the most common, occurring in 35% to 60% of patients. In absence of SLE, DPGN pathology looks more like Membranoproliferative glomerulonephritis
Cryoglobulinemic vasculitis is a form of inflammation affecting the blood vessels caused by the deposition of abnormal proteins called cryoglobulins. These immunoglobulin proteins are soluble at normal body temperatures, but become insoluble below 37 °C (98.6 °F) and subsequently may aggregate within smaller blood vessels. Inflammation within these obstructed blood vessels is due to the deposition of complement proteins which activate inflammatory pathways.
Complement 3 deficiency is a genetic condition affecting complement component 3 (C3). People can suffer from either primary or secondary C3 deficiency. Primary C3 deficiency refers to an inherited autosomal-recessive disorder that involves mutations in the gene for C3. Secondary C3 deficiency results from a lack of factor I or factor H, two proteins that are key for the regulation of C3. Both primary and secondary C3 deficiency are characterized by low levels or absence of C3.
Complement factor H-related protein 5 (CFHR5) nephropathy is a form of inherited kidney disease which is endemic in Cyprus and is caused by a mutation in the gene CFHR5. It is thought to affect up to 1:6000 Cypriots but has not been reported in anybody who is not of Cypriot descent.

Samoyed hereditary glomerulopathy (SHG) is a hereditary, X-linked, noninflammatory disease of the renal glomeruli, occurring in the Samoyed breed of dog. The disease has been shown to be a model for Alport syndrome in humans in that the disease resembles that of the human disease. Because of this, it is sometimes referred to by the name given to the disease in humans when referring to the conditions in Samoyed dogs. Alternatively, it may also be known as X-linked hereditary nephritis. Genetically, the trait is inherited as a sex-linked, genetically dominant disease, and thus affects male dogs to a greater degree than female dogs, since males only have one X chromosome.

Transplant glomerulopathy (TG) is a disease of the glomeruli in transplanted kidneys. It is a type of renal injury often associated with chronic antibody-mediated rejection. However, transplant glomerulopathy is not specific for chronic antibody-mediated rejection; it may be the result of a number of disease processes affecting the glomerular endothelium.
Monoclonal gammopathy of renal significance (MGRS) are a group of kidney disorders that present with kidney damage due to nephrotoxic monoclonal immunoglobulins secreted by clonal plasma cells or B cells. By definition, people with MGRS do not meet criteria for multiple myeloma or other hematologic malignancies. The term MGRS was introduced in 2012 by the International Kidney and Monoclonal Gammopathy Research Group (IKMG). MGRS is associated with monoclonal gammopathy of undetermined significance (MGUS). People with MGUS have a monoclonal gammopathy but does not meet the criteria for the clonal burden nor the presence of end organ damage seen in hematologic malignancies. In a population based study based on the NHANES III health survey; 6% of patients with MGUS were subsequently classified as having MGRS. The prevalence and incidence of MGRS in the general population or in specific populations is not known but it is more prevalent in those over the age of 50 as there is a monoclonal protein (M-protein) present in 3% of those 50 and years older and 5% of those 70 years and older, placing those 50 and older at increased risk of MGRS.

