Humoral immunity is the aspect of immunity that is mediated by macromolecules – including secreted antibodies, complement proteins, and certain antimicrobial peptides – located in extracellular fluids. Humoral immunity is named so because it involves substances found in the humors, or body fluids. It contrasts with cell-mediated immunity. Humoral immunity is also referred to as antibody-mediated immunity.

The complement system, also known as complement cascade, is a part of the humoral, innate immune system and enhances (complements) the ability of antibodies and phagocytic cells to clear microbes and damaged cells from an organism, promote inflammation, and attack the pathogen's cell membrane. Despite being part of the innate immune system, the complement system can be recruited and brought into action by antibodies generated by the adaptive immune system.

The classical complement pathway is one of three pathways which activate the complement system, which is part of the immune system. The classical complement pathway is initiated by antigen-antibody complexes with the antibody isotypes IgG and IgM.

C3 convertase belongs to family of serine proteases and is necessary in innate immunity as a part of the complement system which eventuate in opsonisation of particles, release of inflammatory peptides, C5 convertase formation and cell lysis.

Complement receptor type 1 (CR1) also known as C3b/C4b receptor or CD35 is a protein that in humans is encoded by the CR1 gene.

The membrane attack complex (MAC) or terminal complement complex (TCC) is a complex of proteins typically formed on the surface of pathogen cell membranes as a result of the activation of the host's complement system, and as such is an effector of the immune system. Antibody-mediated complement activation leads to MAC deposition on the surface of infected cells. Assembly of the MAC leads to pores that disrupt the cell membrane of target cells, leading to cell lysis and death.

Complement component 3, often simply called C3, is a protein of the immune system that is found primarily in the blood. It plays a central role in the complement system of vertebrate animals and contributes to innate immunity. In humans it is encoded on chromosome 19 by a gene called C3.

C5 convertase is an enzyme belonging to a family of serine proteases that play key role in the innate immunity. It participates in the complement system ending with cell death.

Properdin is a protein that in humans is encoded by the CFP gene.
Alternative-complement-pathway C3/C5 convertase is an enzyme. This enzyme catalyses the following chemical reaction

Factor D is a protein which in humans is encoded by the CFD gene. Factor D is involved in the alternative complement pathway of the complement system where it cleaves factor B.
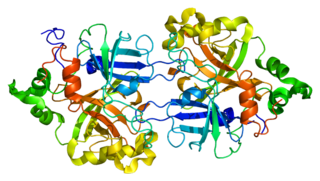
Complement factor B is a protein that in humans is encoded by the CFB gene.
C4b-binding protein (C4BP) is a protein complex involved in the complement system where it acts as inhibitor. C4BP has an octopus-like structure with a central stalk and seven branching alpha-chains. The main form of C4BP in human blood is composed of 7 identical alpha-chains and one unique beta-chain, which in turn binds anticoagulant, vitamin K-dependent protein S.

Complement decay-accelerating factor, also known as CD55 or DAF, is a protein that, in humans, is encoded by the CD55 gene.
Complement control proteins are proteins that interact with components of the complement system.

Factor H (FH) is a member of the regulators of complement activation family and is a complement control protein. It is a large, soluble glycoprotein that circulates in human plasma. Its principal function is to regulate the alternative pathway of the complement system, ensuring that the complement system is directed towards pathogens or other dangerous material and does not damage host tissue. Factor H regulates complement activation on self cells and surfaces by possessing both cofactor activity for Factor I–mediated C3b cleavage, and decay accelerating activity against the alternative pathway C3-convertase, C3bBb. Factor H exerts its protective action on self cells and self surfaces but not on the surfaces of bacteria or viruses. There are however, important exceptions, such as for example the bacterial pathogen, Neisseria meningitidis. This human pathogen has evolved mechanisms to recruit human FH and down-regulate the alternative pathway. Binding of FH permits the bacteria to proliferate in the bloodstream and cause disease.

Complement factor I, also known as C3b/C4b inactivator, is a protein that in humans is encoded by the CFI gene. Complement factor I is a protein of the complement system, first isolated in 1966 in guinea pig serum, that regulates complement activation by cleaving cell-bound or fluid phase C3b and C4b. It is a soluble glycoprotein that circulates in human blood at an average concentration of 35 μg/mL.
C3b is the larger of two elements formed by the cleavage of complement component 3, and is considered an important part of the innate immune system. C3b is potent in opsonization: tagging pathogens, immune complexes (antigen-antibody), and apoptotic cells for phagocytosis. Additionally, C3b plays a role in forming a C3 convertase when bound to Factor B, or a C5 convertase when bound to C4b and C2b or when an additional C3b molecule binds to the C3bBb complex.
C3a is one of the proteins formed by the cleavage of complement component 3; the other is C3b. C3a is a 77 residue anaphylatoxin that binds to the C3a receptor (C3aR), a class A G protein-coupled receptor. It plays a large role in the immune response.
Complement 3 deficiency is a genetic condition affecting complement component 3 (C3). People can suffer from either primary or secondary C3 deficiency. Primary C3 deficiency refers to an inherited autosomal-recessive disorder that involves mutations in the gene for C3. Secondary C3 deficiency results from a lack of factor I or factor H, two proteins that are key for the regulation of C3. Both primary and secondary C3 deficiency are characterized by low levels or absence of C3.

