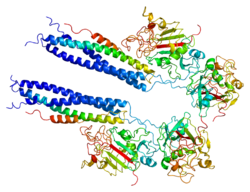




Fibrinogen alpha chain is a protein that in humans is encoded by the FGA gene.
Fibrinogen alpha chain is a protein that in humans is encoded by the FGA gene.
The protein encoded by this gene is the alpha component of fibrinogen, a blood-borne glycoprotein composed of three pairs of nonidentical polypeptide chains. Following vascular injury, fibrinogen is cleaved by thrombin to form fibrin, which is the most abundant component of blood clots. In addition, various cleavage products of fibrinogen and fibrin regulate cell adhesion and spreading, display vasoconstrictor and chemotactic activities, and are mitogens for several cell types. Mutations in this gene lead to several disorders, including dysfibrinogenemia, hypofibrinogenemia, afibrinogenemia, and renal amyloidosis. Alternative splicing results in two isoforms that vary in the carboxy-terminus. [5]
Fibrinogen alpha chain has been shown to interact with tissue plasminogen activator. [6] [7]
PDB gallery | |
|---|---|
|





















