Factor IX is produced as a zymogen, an inactive precursor. It is processed to remove the signal peptide, glycosylated and then cleaved by factor XIa (of the contact pathway) or factor VIIa (of the tissue factor pathway) to produce a two-chain form, where the chains are linked by a disulfide bridge.[7][8] When activated into factor IXa, in the presence of Ca2+, membrane phospholipids, and a Factor VIII cofactor, it hydrolyses one arginine-isoleucine bond in factor X to form factor Xa.
Factor IX expression increases with age in humans and mice. In mouse models, mutations within the promoter region of factor IX have an age-dependent phenotype.[9]
Domain architecture
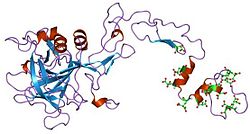
Factors VII, IX, and X all play key roles in blood coagulation and also share a common domain architecture.[10] The factor IX protein is composed of four protein domains: the Gla domain, two tandem copies of the EGF domain and a C-terminal trypsin-like peptidase domain which carries out the catalytic cleavage.
Human factor IX protein domain architecture, where each protein domain is represented by a coloured box
The N-terminal EGF domain has been shown to at least in part be responsible for binding tissue factor.[10] Wilkinson et al. conclude that residues 88 to 109 of the second EGF domain mediate binding to platelets and assembly of the factor X activating complex.[11]
The structures of all four domains have been solved. A structure of the two EGF domains and the trypsin-like domain was determined for the pig protein.[12] The structure of the Gla domain, which is responsible for Ca(II)-dependent phospholipid binding, was also determined by NMR.[13]
Several structures of 'super active' mutants have been solved,[14] which reveal the nature of factor IX activation by other proteins in the clotting cascade.
Genetics
In human, the F9 gene is located on the X chromosome at position q27.1.
Because the gene for factor IX is located on the X chromosome (Xq27.1-q27.2), loss-of-function mutations thereof are X-linked recessive: males experience the disease phenotype much more frequently than females. At least 534 disease-causing mutations in this gene have been discovered.[15] The F9 gene was first cloned in 1982 by Kotoku Kurachi and Earl Davie.[16]
Deficiency of factor IX causes Christmas disease (hemophilia B).[5] Over 3000 variants of factor IX have been described, affecting 73% of the 461 residues;[22] some cause no symptoms, but many lead to a significant bleeding disorder. The original Christmas disease mutation was identified by sequencing of Christmas' DNA, revealing a mutation which changed a cysteine to a serine.[23]Recombinant factor IX is used to treat Christmas disease. Formulations include:
Some rare mutations of factor IX result in elevated clotting activity, and can result in clotting diseases, such as deep vein thrombosis. This gain of function mutation renders the protein hyperfunctional and is associated with familial early-onset thrombophilia.[36]
Factor IX deficiency is treated by injection of purified factor IX produced through cloning in various animal or animal cell vectors. Tranexamic acid may be of value in patients undergoing surgery who have inherited factor IX deficiency in order to reduce the perioperative risk of bleeding.[37]
A list of all the mutations in Factor IX is compiled and maintained by EAHAD.[38]
1 2 World Health Organization (2025). The selection and use of essential medicines, 2025: WHO Model List of Essential Medicines, 24th list. Geneva: World Health Organization. hdl:10665/382243.
↑ Taylor SA, Duffin J, Cameron C, Teitel J, Garvey B, Lillicrap DP (January 1992). "Characterization of the original Christmas disease mutation (cysteine 206----serine): from clinical recognition to molecular pathogenesis". Thrombosis and Haemostasis. 67 (1): 63–5. doi:10.1055/s-0038-1648381. PMID1615485. S2CID25251813.
Lenting PJ, van Mourik JA, Mertens K (December 1998). "The life cycle of coagulation factor VIII in view of its structure and function". Blood. 92 (11): 3983–96. doi:10.1182/blood.V92.11.3983. PMID9834200.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.