
The National Center for Biotechnology Information (NCBI) is part of the United States National Library of Medicine (NLM), a branch of the National Institutes of Health (NIH). It is approved and funded by the government of the United States. The NCBI is located in Bethesda, Maryland, and was founded in 1988 through legislation sponsored by US Congressman Claude Pepper.
In bioinformatics, BLAST is an algorithm and program for comparing primary biological sequence information, such as the amino-acid sequences of proteins or the nucleotides of DNA and/or RNA sequences. A BLAST search enables a researcher to compare a subject protein or nucleotide sequence with a library or database of sequences, and identify database sequences that resemble the query sequence above a certain threshold. For example, following the discovery of a previously unknown gene in the mouse, a scientist will typically perform a BLAST search of the human genome to see if humans carry a similar gene; BLAST will identify sequences in the human genome that resemble the mouse gene based on similarity of sequence.
In the field of bioinformatics, a sequence database is a type of biological database that is composed of a large collection of computerized ("digital") nucleic acid sequences, protein sequences, or other polymer sequences stored on a computer. The UniProt database is an example of a protein sequence database. As of 2013 it contained over 40 million sequences and is growing at an exponential rate. Historically, sequences were published in paper form, but as the number of sequences grew, this storage method became unsustainable.

Biological databases are libraries of biological sciences, collected from scientific experiments, published literature, high-throughput experiment technology, and computational analysis. They contain information from research areas including genomics, proteomics, metabolomics, microarray gene expression, and phylogenetics. Information contained in biological databases includes gene function, structure, localization, clinical effects of mutations as well as similarities of biological sequences and structures.

The Entrez Global Query Cross-Database Search System is a federated search engine, or web portal that allows users to search many discrete health sciences databases at the National Center for Biotechnology Information (NCBI) website. The NCBI is a part of the National Library of Medicine (NLM), which is itself a department of the National Institutes of Health (NIH), which in turn is a part of the United States Department of Health and Human Services. The name "Entrez" was chosen to reflect the spirit of welcoming the public to search the content available from the NLM.
In computational biology, gene prediction or gene finding refers to the process of identifying the regions of genomic DNA that encode genes. This includes protein-coding genes as well as RNA genes, but may also include prediction of other functional elements such as regulatory regions. Gene finding is one of the first and most important steps in understanding the genome of a species once it has been sequenced.
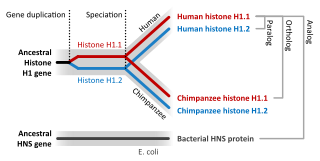
Sequence homology is the biological homology between DNA, RNA, or protein sequences, defined in terms of shared ancestry in the evolutionary history of life. Two segments of DNA can have shared ancestry because of three phenomena: either a speciation event (orthologs), or a duplication event (paralogs), or else a horizontal gene transfer event (xenologs).
The Rat Genome Database (RGD) is a database of rat genomics, genetics, physiology and functional data, as well as data for comparative genomics between rat, human and mouse. RGD is responsible for attaching biological information to the rat genome via structured vocabulary, or ontology, annotations assigned to genes and quantitative trait loci (QTL), and for consolidating rat strain data and making it available to the research community. They are also developing a suite of tools for mining and analyzing genomic, physiologic and functional data for the rat, and comparative data for rat, mouse, human, and five other species.
The Bioinformatic Harvester was a bioinformatic meta search engine created by the European Molecular Biology Laboratory and subsequently hosted and further developed by KIT Karlsruhe Institute of Technology for genes and protein-associated information. Harvester currently works for human, mouse, rat, zebrafish, drosophila and arabidopsis thaliana based information. Harvester cross-links >50 popular bioinformatic resources and allows cross searches. Harvester serves tens of thousands of pages every day to scientists and physicians. Since 2014 the service is down.
The completion of the human genome sequencing in the early 2000s was a turning point in genomics research. Scientists have conducted series of research into the activities of genes and the genome as a whole. The human genome contains around 3 billion base pairs nucleotide, and the huge quantity of data created necessitates the development of an accessible tool to explore and interpret this information in order to investigate the genetic basis of disease, evolution, and biological processes. The field of genomics has continued to grow, with new sequencing technologies and computational tool making it easier to study the genome.
The Saccharomyces Genome Database (SGD) is a scientific database of the molecular biology and genetics of the yeast Saccharomyces cerevisiae, which is commonly known as baker's or budding yeast. Further information is located at the Yeastract curated repository.
The Biomolecular Object Network Databank is a bioinformatics databank containing information on small molecule structures and interactions. The databank integrates a number of existing databases to provide a comprehensive overview of the information currently available for a given molecule.
Mouse Genome Informatics (MGI) is a free, online database and bioinformatics resource hosted by The Jackson Laboratory, with funding by the National Human Genome Research Institute (NHGRI), the National Cancer Institute (NCI), and the Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD). MGI provides access to data on the genetics, genomics and biology of the laboratory mouse to facilitate the study of human health and disease. The database integrates multiple projects, with the two largest contributions coming from the Mouse Genome Database and Mouse Gene Expression Database (GXD). As of 2018, MGI contains data curated from over 230,000 publications.
UniGene was a NCBI database of the transcriptome and thus, despite the name, not primarily a database for genes. Each entry is a set of transcripts that appear to stem from the same transcription locus. Information on protein similarities, gene expression, cDNA clones, and genomic location is included with each entry.
The Reference Sequence (RefSeq) database is an open access, annotated and curated collection of publicly available nucleotide sequences and their protein products. RefSeq was introduced in 2000. This database is built by National Center for Biotechnology Information (NCBI), and, unlike GenBank, provides only a single record for each natural biological molecule for major organisms ranging from viruses to bacteria to eukaryotes.

The Genome Reference Consortium (GRC) is an international collective of academic and research institutes with expertise in genome mapping, sequencing, and informatics, formed to improve the representation of reference genomes. At the time the human reference was initially described, it was clear that some regions were recalcitrant to analysis with existing technology, leaving gaps in the known sequence. The main reason for improving the reference assemblies are that they are the cornerstones upon which all whole genome studies are based.
GeneCards is a database of human genes, which provides genomic, proteomic, transcriptomic, genetic, medical, and functional information on all known and predicted human genes. It is being developed and maintained by the Crown Human Genome Center at the Weizmann Institute of Science, in collaboration with LifeMap Sciences.

Family with Sequence Similarity 203, Member B (FAM203B) is a protein encoded by the FAM203B gene (8q24.3) in humans. While FAM203B is only found in humans and possibly non-human primates, its paralog, FAM203A, is highly conserved. The FAM203B protein contains two conserved domains of unknown function, DUF383 and DUF384, and no transmembrane domains. This protein has no known function yet, although the homolog of FAM203A in Caenorhabditis elegans (Y54H5A.2) is thought to help regulate the actin cytoskeleton.

MIPOL1 , also known as CCDC193 , is a protein that in humans is encoded by the MIPOL1 gene. Mutation of this gene is associated with mirror-image polydactyly in humans, which is a rare genetic condition characterized by mirror-image duplication of digits.