Conversion of a gene's sequence into a mature gene product or products
Gene expression is the process by which the information contained within a gene is used to produce a functional gene product, such as a protein or a functional RNA molecule. This process involves multiple steps, including the transcription of the gene's sequence into RNA. For protein-coding genes, this RNA is further translated into a chain of amino acids that folds into a protein, while for non-coding genes, the resulting RNA itself serves a functional role in the cell. Gene expression enables cells to utilize the genetic information in genes to carry out a wide range of biological functions. While expression levels can be regulated in response to cellular needs and environmental changes, some genes are expressed continuously with little variation.[1]
The production of a RNA copy from a DNA strand is called transcription, and is performed by RNA polymerases, which add one ribonucleotide at a time to a growing RNA strand as per the complementarity law of the nucleotide bases. This RNA is complementary to the template 3′ → 5′ DNA strand,[2] with the exception that thymines (T) are replaced with uracils (U) in the RNA and possible errors.
In bacteriatranscription is carried out by a single type of RNA polymerase, which needs to bind a DNA sequence called a Pribnow box with the help of the sigma factor protein (σ factor) to start transcription. In eukaryotes, transcription is performed in the nucleus by three types of RNA polymerases, each of which needs a special DNA sequence called the promoter and a set of DNA-binding proteins—transcription factors—to initiate the process (see regulation of transcription below). RNA polymerase I is responsible for transcription of ribosomal RNA (rRNA) genes. RNA polymerase II (Pol II) transcribes all protein-coding genes but also some non-coding RNAs (e.g., snRNAs, snoRNAs or long non-coding RNAs). RNA polymerase III transcribes 5S rRNA, transfer RNA (tRNA) genes, and some small non-coding RNAs (e.g., 7SK). Transcription ends when the polymerase encounters a sequence called the terminator.
While transcription of prokaryotic protein-coding genes creates messenger RNA (mRNA) that is ready for translation into protein, transcription of eukaryotic genes leaves a primary transcript of RNA (pre-RNA), which first has to undergo a series of modifications to become a mature RNA. Types and steps involved in the maturation processes vary between coding and non-coding preRNAs; i.e. even though preRNA molecules for both mRNA and tRNA undergo splicing, the steps and machinery involved are different.[3] The processing of non-coding RNA is described below (non-coding RNA maturation).
The processing of pre-mRNA include 5′ capping, which is set of enzymatic reactions that add 7-methylguanosine (m7G) to the 5′ end of pre-mRNA and thus protect the RNA from degradation by exonucleases.[4] The m7G cap is then bound by cap binding complex heterodimer (CBP20/CBP80), which aids in mRNA export to cytoplasm and also protect the RNA from decapping.[5]
Another modification is 3′ cleavage and polyadenylation.[6] They occur if polyadenylation signal sequence (5′- AAUAAA-3′) is present in pre-mRNA, which is usually between protein-coding sequence and terminator.[7] The pre-mRNA is first cleaved and then a series of ~200 adenines (A) are added to form poly(A) tail, which protects the RNA from degradation.[8] The poly(A) tail is bound by multiple poly(A)-binding proteins (PABPs) necessary for mRNA export and translation re-initiation.[9] In the inverse process of deadenylation, poly(A) tails are shortened by the CCR4-Not 3′-5′ exonuclease, which often leads to full transcript decay.[10]
Illustration of exons and introns in pre-mRNA and the formation of mature mRNA by splicing. The UTRs (in green) are non-coding parts of exons at the ends of the mRNA.
A very important modification of eukaryotic pre-mRNA is RNA splicing. The majority of eukaryotic pre-mRNAs consist of alternating segments called exons and introns.[11] During the process of splicing, an RNA-protein catalytical complex known as spliceosome catalyzes two transesterification reactions, which remove an intron and release it in form of lariat structure, and then splice neighbouring exons together.[12] In certain cases, some introns or exons can be either removed or retained in mature mRNA.[13] This so-called alternative splicing creates series of different transcripts originating from a single gene. Because these transcripts can be potentially translated into different proteins, splicing extends the complexity of eukaryotic gene expression and the size of a species proteome.[14]
Extensive RNA processing may be an evolutionary advantage made possible by the nucleus of eukaryotes. In prokaryotes, transcription and translation happen together, whilst in eukaryotes, the nuclear membrane separates the two processes, giving time for RNA processing to occur.[15]
In most organisms non-coding genes (ncRNA) are transcribed as precursors that undergo further processing. In the case of ribosomal RNAs (rRNA), they are often transcribed as a pre-rRNA that contains one or more rRNAs. The pre-rRNA is cleaved and modified (2′-O-methylation and pseudouridine formation) at specific sites by approximately 150 different small nucleolus-restricted RNA species, called snoRNAs. SnoRNAs associate with proteins, forming snoRNPs. While snoRNA part basepair with the target RNA and thus position the modification at a precise site, the protein part performs the catalytical reaction. In eukaryotes, in particular a snoRNP called RNase, MRP cleaves the 45S pre-rRNA into the 28S, 5.8S, and 18S rRNAs. The rRNA and RNA processing factors form large aggregates called the nucleolus.[16]
In the case of transfer RNA (tRNA), for example, the 5′ sequence is removed by RNase P,[17] whereas the 3′ end is removed by the tRNase Z enzyme[18] and the non-templated 3′ CCA tail is added by a nucleotidyl transferase.[19] In the case of micro RNA (miRNA), miRNAs are first transcribed as primary transcripts or pri-miRNA with a cap and poly-A tail and processed to short, 70-nucleotide stem-loop structures known as pre-miRNA in the cell nucleus by the enzymes Drosha and Pasha. After being exported, it is then processed to mature miRNAs in the cytoplasm by interaction with the endonuclease Dicer, which also initiates the formation of the RNA-induced silencing complex (RISC), composed of the Argonaute protein.
Even snRNAs and snoRNAs themselves undergo series of modification before they become part of functional RNP complex.[20] This is done either in the nucleoplasm or in the specialized compartments called Cajal bodies.[21] Their bases are methylated or pseudouridinilated by a group of small Cajal body-specific RNAs (scaRNAs), which are structurally similar to snoRNAs.[22]
For some non-coding RNA, the mature RNA is the final gene product.[23] In the case of messenger RNA (mRNA) the RNA is an information carrier coding for the synthesis of one or more proteins. mRNA carrying a single protein sequence (common in eukaryotes) is monocistronic whilst mRNA carrying multiple protein sequences (common in prokaryotes) is known as polycistronic.
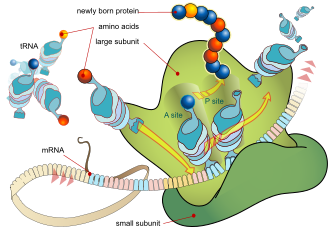
During the translation, tRNA charged with amino acid enters the ribosome and aligns with the correct mRNA triplet. Ribosome then adds amino acid to growing protein chain.
Every mRNA consists of three parts: a 5′ untranslated region (5′UTR), a protein-coding region or open reading frame (ORF), and a 3′ untranslated region (3′UTR). The coding region carries information for protein synthesis encoded by the genetic code to form triplets. Each triplet of nucleotides of the coding region is called a codon and corresponds to a binding site complementary to an anticodon triplet in transfer RNA. Transfer RNAs with the same anticodon sequence always carry an identical type of amino acid. Amino acids are then chained together by the ribosome according to the order of triplets in the coding region. The ribosome helps transfer RNA to bind to messenger RNA and takes the amino acid from each transfer RNA and makes a structure-less protein out of it.[24][25] Each mRNA molecule is translated into many protein molecules, on average ~2800 in mammals.[26][27]
In prokaryotes translation generally occurs at the point of transcription (co-transcriptionally), often using a messenger RNA that is still in the process of being created. In eukaryotes translation can occur in a variety of regions of the cell depending on where the protein being written is supposed to be. Major locations are the cytoplasm for soluble cytoplasmic proteins and the membrane of the endoplasmic reticulum for proteins that are for export from the cell or insertion into a cell membrane. Proteins that are supposed to be produced at the endoplasmic reticulum are recognised part-way through the translation process. This is governed by the signal recognition particle—a protein that binds to the ribosome and directs it to the endoplasmic reticulum when it finds a signal peptide on the growing (nascent) amino acid chain.[28]
The patchy colours of a tortoiseshell cat are the result of different levels of expression of pigmentation genes in different areas of the skin.
Regulation of gene expression is the control of the amount and timing of appearance of the functional product of a gene. Control of expression is vital to allow a cell to produce the gene products it needs when it needs them; in turn, this gives cells the flexibility to adapt to a variable environment, external signals, damage to the cell, and other stimuli. More generally, gene regulation gives the cell control over all structure and function, and is the basis for cellular differentiation, morphogenesis and the versatility and adaptability of any organism.
Numerous terms are used to describe types of genes depending on how they are regulated; these include:
A constitutive gene is a gene that is transcribed continually as opposed to a facultative gene, which is only transcribed when needed.
A housekeeping gene is a gene that is required to maintain basic cellular function and so is typically expressed in all cell types of an organism. Examples include actin, GAPDH and ubiquitin. Some housekeeping genes are transcribed at a relatively constant rate and these genes can be used as a reference point in experiments to measure the expression rates of other genes.
A facultative gene is a gene only transcribed when needed as opposed to a constitutive gene.
An inducible gene is a gene whose expression is either responsive to environmental change or dependent on the position in the cell cycle.
Any step of gene expression may be modulated, from the DNA-RNA transcription step to post-translational modification of a protein. The stability of the final gene product, whether it is RNA or protein, also contributes to the expression level of the gene—an unstable product results in a low expression level. In general gene expression is regulated through changes[29] in the number and type of interactions between molecules[30] that collectively influence transcription of DNA[31] and translation of RNA.[32]
Some simple examples of where gene expression is important are:
When lactose is present in a prokaryote, it acts as an inducer and inactivates the repressor so that the genes for lactose metabolism can be transcribed.
Regulation of transcription can be broken down into three main routes of influence; genetic (direct interaction of a control factor with the gene), modulation interaction of a control factor with the transcription machinery and epigenetic (non-sequence changes in DNA structure that influence transcription).[33][34]
The lambda repressor transcription factor (green) binds as a dimer to major groove of DNA target (red and blue) and disables initiation of transcription. From PDB: 1LMB.
Direct interaction with DNA is the simplest and the most direct method by which a protein changes transcription levels.[35] Genes often have several protein binding sites around the coding region with the specific function of regulating transcription.[36] There are many classes of regulatory DNA binding sites known as enhancers, insulators and silencers.[37] The mechanisms for regulating transcription are varied, from blocking key binding sites on the DNA for RNA polymerase to acting as an activator and promoting transcription by assisting RNA polymerase binding.[38]
The activity of transcription factors is further modulated by intracellular signals causing protein post-translational modification including phosphorylation, acetylation, or glycosylation.[39] These changes influence a transcription factor's ability to bind, directly or indirectly, to promoter DNA, to recruit RNA polymerase, or to favor elongation of a newly synthesized RNA molecule.[40]
The nuclear membrane in eukaryotes allows further regulation of transcription factors by the duration of their presence in the nucleus, which is regulated by reversible changes in their structure and by binding of other proteins.[41] Environmental stimuli or endocrine signals[42] may cause modification of regulatory proteins[43] eliciting cascades of intracellular signals,[44] which result in regulation of gene expression.
It has become apparent that there is a significant influence of non-DNA-sequence specific effects on transcription.[45] These effects are referred to as epigenetic and involve the higher order structure of DNA, non-sequence specific DNA binding proteins and chemical modification of DNA.[46] In general epigenetic effects alter the accessibility of DNA to proteins and so modulate transcription.[47]
In eukaryotes, DNA is organized in form of nucleosomes. Note how the DNA (blue and green) is tightly wrapped around the protein core made of histoneoctamer (ribbon coils), restricting access to the DNA. From PDB: 1KX5.
Enhancers, transcription factors, mediator complex and DNA loops
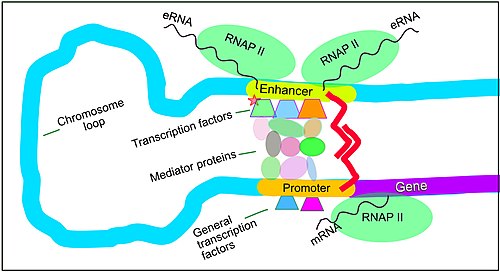
Regulation of transcription in mammals. An active enhancer regulatory region is enabled to interact with the promoter region of its target gene by formation of a chromosome loop. This can initiate messenger RNA (mRNA) synthesis by RNA polymerase II (RNAP II) bound to the promoter at the transcription start site of the gene. The loop is stabilized by one architectural protein anchored to the enhancer and one anchored to the promoter and these proteins are joined to form a dimer (red zigzags). Specific regulatory transcription factors bind to DNA sequence motifs on the enhancer. General transcription factors bind to the promoter. When a transcription factor is activated by a signal (here indicated as phosphorylation shown by a small red star on a transcription factor on the enhancer) the enhancer is activated and can now activate its target promoter. The active enhancer is transcribed on each strand of DNA in opposite directions by bound RNAP IIs. Mediator proteins (a complex consisting of about 26 proteins in an interacting structure) communicate regulatory signals from the enhancer DNA-bound transcription factors to the promoter.
Enhancers are genome regions that regulate genes. Enhancers control cell-type-specific gene expression programs, most often by looping through long distances to come in physical proximity with the promoters of their target genes.[51] Multiple enhancers, each often tens or hundred of thousands of nucleotides distant from their target genes, loop to their target gene promoters and coordinate with each other to control gene expression.[51]
The illustration shows an enhancer looping around to come into proximity with the promoter of a target gene. The loop is stabilized by a dimer of a connector protein (e.g. dimer of CTCF or YY1). One member of the dimer is anchored to its binding motif on the enhancer and the other member is anchored to its binding motif on the promoter (represented by the red zigzags in the illustration).[52] Several cell function-specific transcription factors (among the about 1,600 transcription factors in a human cell)[53] generally bind to specific motifs on an enhancer.[54] A small combination of these enhancer-bound transcription factors, when brought close to a promoter by a DNA loop, govern transcription level of the target gene. Mediator (a complex usually consisting of about 26 proteins in an interacting structure) communicates regulatory signals from enhancer DNA-bound transcription factors directly to the RNA polymerase II (pol II) enzyme bound to the promoter.[55]
Enhancers, when active, are generally transcribed from both strands of DNA with RNA polymerases acting in two different directions, producing two eRNAs as illustrated in the figure.[56] An inactive enhancer may be bound by an inactive transcription factor. Phosphorylation of the transcription factor may activate it and that activated transcription factor may then activate the enhancer to which it is bound (see small red star representing phosphorylation of transcription factor bound to enhancer in the illustration).[57] An activated enhancer begins transcription of its RNA before activating transcription of messenger RNA from its target gene.[58]
DNA methylation and demethylation
DNA methylation is the addition of a methyl group to the DNA that happens at cytosine. The image shows a cytosine single ring base and a methyl group added on to the 5 carbon. In mammals, DNA methylation occurs almost exclusively at a cytosine that is followed by a guanine.
DNA methylation is a widespread mechanism for epigenetic influence on gene expression and is seen in bacteria and eukaryotes and has roles in heritable transcription silencing and transcription regulation. Methylation most often occurs on a cytosine (see Figure). Methylation of cytosine primarily occurs in dinucleotide sequences where a cytosine is followed by a guanine, a CpG site. The number of CpG sites in the human genome is about 28 million.[59] Depending on the type of cell, about 70% of the CpG sites have a methylated cytosine.[60]
Methylation of cytosine in DNA has a major role in regulating gene expression. Methylation of CpGs in a promoter region of a gene usually represses gene transcription[61] while methylation of CpGs in the body of a gene increases expression.[62]TET enzymes play a central role in demethylation of methylated cytosines. Demethylation of CpGs in a gene promoter by TET enzyme activity increases transcription of the gene.[63]
In eukaryotes, where export of RNA is required before translation is possible, nuclear export is thought to provide additional control over gene expression. All transport in and out of the nucleus is via the nuclear pore and transport is controlled by a wide range of importin and exportin proteins.[64]
Expression of a gene coding for a protein is only possible if the messenger RNA carrying the code survives long enough to be translated.[65] In a typical cell, an RNA molecule is only stable if specifically protected from degradation.[66] RNA degradation has particular importance in regulation of expression in eukaryotic cells where mRNA has to travel significant distances before being translated.[67] In eukaryotes, RNA is stabilised by certain post-transcriptional modifications, particularly the 5′ cap and poly-adenylated tail.[9]
Intentional degradation of mRNA is used not just as a defence mechanism from foreign RNA (normally from viruses) but also as a route of mRNA destabilisation.[68] If an mRNA molecule has a complementary sequence to a small interfering RNA then it is targeted for destruction via the RNA interference pathway.[69]
Three prime untranslated regions (3′UTRs) of messenger RNAs (mRNAs) often contain regulatory sequences that post-transcriptionally influence gene expression. Such 3′-UTRs often contain both binding sites for microRNAs (miRNAs) as well as for regulatory proteins.[70] By binding to specific sites within the 3′-UTR, miRNAs can decrease gene expression of various mRNAs by either inhibiting translation or directly causing degradation of the transcript.[71] The 3′-UTR also may have silencer regions that bind repressor proteins that inhibit the expression of a mRNA.[72]
The 3′-UTR often contains microRNA response elements (MREs). MREs are sequences to which miRNAs bind. These are prevalent motifs within 3′-UTRs. Among all regulatory motifs within the 3′-UTRs (e.g. including silencer regions), MREs make up about half of the motifs.[73]
As of 2014, the miRBase web site,[74] an archive of miRNAsequences and annotations, listed 28,645 entries in 233 biologic species. Of these, 1,881 miRNAs were in annotated human miRNA loci. miRNAs were predicted to have an average of about four hundred target mRNAs (affecting expression of several hundred genes).[75] Friedman et al.[75] estimate that >45,000 miRNA target sites within human mRNA 3′UTRs are conserved above background levels, and >60% of human protein-coding genes have been under selective pressure to maintain pairing to miRNAs.
Direct experiments show that a single miRNA can reduce the stability of hundreds of unique mRNAs.[76] Other experiments show that a single miRNA may repress the production of hundreds of proteins, but that this repression often is relatively mild (less than 2-fold).[77][78]
The effects of miRNA dysregulation of gene expression seem to be important in cancer.[79] For instance, in gastrointestinal cancers, nine miRNAs have been identified as epigenetically altered and effective in down regulating DNA repair enzymes.[80]
The effects of miRNA dysregulation of gene expression also seem to be important in neuropsychiatric disorders, such as schizophrenia, bipolar disorder, major depression, Parkinson's disease, Alzheimer's disease and autism spectrum disorders.[81][82]
Translational
Neomycin is an example of a small molecule that reduces expression of all protein genes inevitably leading to cell death; it thus acts as an antibiotic.
Direct regulation of translation is less prevalent than control of transcription or mRNA stability but is occasionally used.[83] Inhibition of protein translation is a major target for toxins and antibiotics, so they can kill a cell by overriding its normal gene expression control.[84]Protein synthesis inhibitors include the antibiotic neomycin and the toxin ricin.[85]
Post-translational modifications (PTMs) are covalent modifications to proteins. Like RNA splicing, they help to significantly diversify the proteome. These modifications are usually catalyzed by enzymes. Additionally, processes like covalent additions to amino acid side chain residues can often be reversed by other enzymes. However, some, like the proteolytic cleavage of the protein backbone, are irreversible.[86]
PTMs play many important roles in the cell.[87] For example, phosphorylation is primarily involved in activating and deactivating proteins and in signaling pathways.[88] PTMs are involved in transcriptional regulation: an important function of acetylation and methylation is histone tail modification, which alters how accessible DNA is for transcription.[86] They can also be seen in the immune system, where glycosylation plays a key role.[89] One type of PTM can initiate another type of PTM, as can be seen in how ubiquitination tags proteins for degradation through proteolysis.[86] Proteolysis, other than being involved in breaking down proteins, is also important in activating and deactivating them, and in regulating biological processes such as DNA transcription and cell death.[90]
Measurement
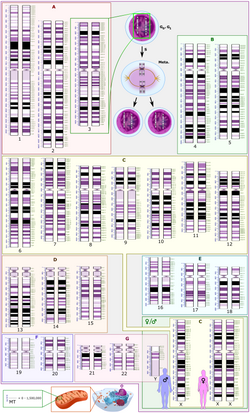
Schematic karyogram of a human, showing an overview of the expression of the human genome using G banding, which is a method that includes Giemsa staining, wherein the lighter staining regions are generally more transcriptionally active, whereas darker regions are more inactive.
Measuring gene expression is an important part of many life sciences, as the ability to quantify the level at which a particular gene is expressed within a cell, tissue or organism can provide a lot of valuable information. For example, measuring gene expression can:
Identify viral infection of a cell (viral protein expression).
Determine an individual's susceptibility to cancer (oncogene expression).
Similarly, the analysis of the location of protein expression is a powerful tool, and this can be done on an organismal or cellular scale. Investigation of localization is particularly important for the study of development in multicellular organisms and as an indicator of protein function in single cells. Ideally, measurement of expression is done by detecting the final gene product (for many genes, this is the protein); however, it is often easier to detect one of the precursors, typically mRNA and to infer gene-expression levels from these measurements.
mRNA quantification
Levels of mRNA can be quantitatively measured by northern blotting, which provides size and sequence information about the mRNA molecules.[91] A sample of RNA is separated on an agarose gel and hybridized to a radioactively labeled RNA probe that is complementary to the target sequence.[92] The radiolabeled RNA is then detected by an autoradiograph.[93] Because the use of radioactive reagents makes the procedure time-consuming and potentially dangerous, alternative labeling and detection methods, such as digoxigenin and biotin chemistries, have been developed.[94] Perceived disadvantages of Northern blotting are that large quantities of RNA are required and that quantification may not be completely accurate, as it involves measuring band strength in an image of a gel.[95] On the other hand, the additional mRNA size information from the Northern blot allows the discrimination of alternately spliced transcripts.[96][97]
Another approach for measuring mRNA abundance is RT-qPCR. In this technique, reverse transcription is followed by quantitative PCR. Reverse transcription first generates a DNA template from the mRNA; this single-stranded template is called cDNA. The cDNA template is then amplified in the quantitative step, during which the fluorescence emitted by labeled hybridization probes or intercalating dyes changes as the DNA amplification process progresses.[98] With a carefully constructed standard curve, qPCR can produce an absolute measurement of the number of copies of original mRNA, typically in units of copies per nanolitre of homogenized tissue or copies per cell.[99] qPCR is very sensitive (detection of a single mRNA molecule is theoretically possible), but can be expensive depending on the type of reporter used; fluorescently labeled oligonucleotide probes are more expensive than non-specific intercalating fluorescent dyes.[100]
For expression profiling, or high-throughput analysis of many genes within a sample, quantitative PCR may be performed for hundreds of genes simultaneously in the case of low-density arrays.[101] A second approach is the hybridization microarray. A single array or "chip" may contain probes to determine transcript levels for every known gene in the genome of one or more organisms.[102] Alternatively, "tag based" technologies like Serial analysis of gene expression (SAGE) and RNA-Seq, which can provide a relative measure of the cellular concentration of different mRNAs, can be used.[103] An advantage of tag-based methods is the "open architecture", allowing for the exact measurement of any transcript, with a known or unknown sequence.[104] Next-generation sequencing (NGS) such as RNA-Seq is another approach, producing vast quantities of sequence data that can be matched to a reference genome. Although NGS is comparatively time-consuming, expensive, and resource-intensive, it can identify single-nucleotide polymorphisms, splice-variants, and novel genes, and can also be used to profile expression in organisms for which little or no sequence information is available.[105]
Protein quantification
For genes encoding proteins, the expression level can be directly assessed by a number of methods with some clear analogies to the techniques for mRNA quantification.
One of the most commonly used methods is to perform a Western blot against the protein of interest.[106] This gives information on the size of the protein in addition to its identity. A sample (often cellular lysate) is separated on a polyacrylamide gel, transferred to a membrane and then probed with an antibody to the protein of interest. The antibody can either be conjugated to a fluorophore or to horseradish peroxidase for imaging and/or quantification. The gel-based nature of this assay makes quantification less accurate, but it has the advantage of being able to identify later modifications to the protein, for example proteolysis or ubiquitination, from changes in size.
mRNA-protein correlation
While transcription directly reflects gene expression, the copy number of mRNA molecules does not directly correlate with the number of protein molecules translated from mRNA. Quantification of both protein and mRNA permits a correlation of the two levels. Regulation on each step of gene expression can impact the correlation, as shown for regulation of translation[27] or protein stability.[107] Post-translational factors, such as protein transport in highly polar cells,[108] can influence the measured mRNA-protein correlation as well.
In situ-hybridization of Drosophilaembryos at different developmental stages for the mRNA responsible for the expression of hunchback. High intensity of blue color marks places with high hunchback mRNA quantity.
Analysis of expression is not limited to quantification; localization can also be determined. mRNA can be detected with a suitably labelled complementary mRNA strand and protein can be detected via labelled antibodies. The probed sample is then observed by microscopy to identify where the mRNA or protein is.
The three-dimensional structure of green fluorescent protein. The residues in the centre of the "barrel" are responsible for production of green light after exposing to higher energetic blue light. From PDB: 1EMA.
By replacing the gene with a new version fused to a green fluorescent protein marker or similar, expression may be directly quantified in live cells. This is done by imaging using a fluorescence microscope. It is very difficult to clone a GFP-fused protein into its native location in the genome without affecting expression levels, so this method often cannot be used to measure endogenous gene expression. It is, however, widely used to measure the expression of a gene artificially introduced into the cell, for example via an expression vector. By fusing a target protein to a fluorescent reporter, the protein's behavior, including its cellular localization and expression level, can be significantly changed.
The enzyme-linked immunosorbent assay works by using antibodies immobilised on a microtiter plate to capture proteins of interest from samples added to the well. Using a detection antibody conjugated to an enzyme or fluorophore the quantity of bound protein can be accurately measured by fluorometric or colourimetric detection. The detection process is very similar to that of a Western blot, but by avoiding the gel steps more accurate quantification can be achieved.
↑Krebs JE, Goldstein ES, Kilpatrick ST (2017-03-02). Lewin's genes XII. Burlington, MA: Jones & Bartlett Learning. ISBN978-1-284-10449-3. OCLC965781334.
↑Berk V, Cate JH (June 2007). "Insights into protein biosynthesis from structures of bacterial ribosomes". Current Opinion in Structural Biology. 17 (3): 302–309. doi:10.1016/j.sbi.2007.05.009. PMID17574829.
↑Veitia RA (November 2008). "One thousand and one ways of making functionally similar transcriptional enhancers". BioEssays. 30 (11–12): 1052–1057. doi:10.1002/bies.20849. PMID18937349.
↑Paul S (November 2008). "Dysfunction of the ubiquitin-proteasome system in multiple disease conditions: therapeutic approaches". BioEssays. 30 (11–12): 1172–1184. doi:10.1002/bies.20852. PMID18937370. S2CID29422790.
↑Weber M, Hellmann I, Stadler MB, Ramos L, Pääbo S, Rebhan M, etal. (April 2007). "Distribution, silencing potential and evolutionary impact of promoter DNA methylation in the human genome". Nature Genetics. 39 (4): 457–466. doi:10.1038/ng1990. PMID17334365. S2CID22446734.
↑Alberts B, Johnson A, Lewis J, Raff M, Roberts K, Walter P (2002), "From RNA to Protein", Molecular Biology of the Cell. 4th edition, Garland Science, retrieved 2024-06-10
↑Boyle A, Perry-O'Keefe H (May 2001). "Labeling and colorimetric detection of nonisotopic probes". Current Protocols in Molecular Biology. 3 (Supplement 20): Unit3.18. doi:10.1002/0471142727.mb0318s20. PMID18265226.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.