Regulation of gene expression by a hormone receptorDiagram showing at which stages in the DNA-mRNA-protein pathway expression can be controlled
Regulation of gene expression, or gene regulation,[1] includes a wide range of mechanisms that are used by cells to increase or decrease the production of specific gene products (protein or RNA). Sophisticated programs of gene expression are widely observed in biology, for example to trigger developmental pathways, respond to environmental stimuli, or adapt to new food sources. Virtually any step of gene expression can be modulated, from transcriptional initiation, to RNA processing, and to the post-translational modification of a protein. Often, one gene regulator controls another, and so on, in a gene regulatory network.
Gene regulation is essential for viruses, prokaryotes and eukaryotes as it increases the versatility and adaptability of an organism by allowing the cell to express protein when needed. Although as early as 1951, Barbara McClintock showed interaction between two genetic loci, Activator (Ac) and Dissociator (Ds), in the color formation of maize seeds, the first discovery of a gene regulation system is widely considered to be the identification in 1961 of the lac operon, discovered by François Jacob and Jacques Monod, in which some enzymes involved in lactose metabolism are expressed by E. coli only in the presence of lactose and absence of glucose.
In multicellular organisms, gene regulation drives cellular differentiation and morphogenesis in the embryo, leading to the creation of different cell types that possess different gene expression profiles from the same genome sequence. Although this does not explain how gene regulation originated, evolutionary biologists include it as a partial explanation of how evolution works at a molecular level, and it is central to the science of evolutionary developmental biology ("evo-devo").
Regulated stages of gene expression
Any step of gene expression may be modulated, from signaling to transcription to post-translational modification of a protein. The following is a list of stages where gene expression is regulated, where the most extensively utilized point is transcription initiation, the first stage in transcription:[citation needed]
Histone tails and their function in chromatin formation
In eukaryotes, the accessibility of large regions of DNA can depend on its chromatin structure, which can be altered as a result of histone modifications directed by DNA methylation, ncRNA, or DNA-binding protein. Hence these modifications may up or down regulate the expression of a gene. Some of these modifications that regulate gene expression are inheritable and are referred to as epigenetic regulation.[citation needed]
Structural
Transcription of DNA is dictated by its structure. In general, the density of its packing is indicative of the frequency of transcription. Octameric protein complexes called histones together with a segment of DNA wound around the eight histone proteins (together referred to as a nucleosome) are responsible for the amount of supercoiling of DNA, and these complexes can be temporarily modified by processes such as phosphorylation or more permanently modified by processes such as methylation. Such modifications are considered to be responsible for more or less permanent changes in gene expression levels.[2]
Chemical
Methylation of DNA is a common method of gene silencing. DNA is typically methylated by methyltransferase enzymes on cytosine nucleotides in a CpG dinucleotide sequence (also called "CpG islands" when densely clustered). Analysis of the pattern of methylation in a given region of DNA (which can be a promoter) can be achieved through a method called bisulfite mapping. Methylated cytosine residues are unchanged by the treatment, whereas unmethylated ones are changed to uracil. The differences are analyzed by DNA sequencing or by methods developed to quantify SNPs, such as Pyrosequencing (Biotage) or MassArray (Sequenom), measuring the relative amounts of C/T at the CG dinucleotide. Abnormal methylation patterns are thought to be involved in oncogenesis.[3]
Histone acetylation is also an important process in transcription. Histone acetyltransferase enzymes (HATs) such as CREB-binding protein also dissociate the DNA from the histone complex, allowing transcription to proceed. Often, DNA methylation and histone deacetylation work together in gene silencing. The combination of the two seems to be a signal for DNA to be packed more densely, lowering gene expression.[citation needed]
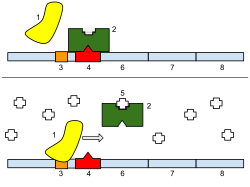
1: RNA Polymerase, 2: Repressor, 3: Promoter, 4: Operator, 5: Lactose, 6: lacZ, 7: lacY, 8: lacA. Top: The gene is essentially turned off. There is no lactose to inhibit the repressor, so the repressor binds to the operator, which obstructs the RNA polymerase from binding to the promoter and making lactase. Bottom: The gene is turned on. Lactose is inhibiting the repressor, allowing the RNA polymerase to bind with the promoter, and express the genes, which synthesize lactase. Eventually, the lactase will digest all of the lactose, until there is none to bind to the repressor. The repressor will then bind to the operator, stopping the manufacture of lactase.
Regulation of transcription thus controls when transcription occurs and how much RNA is created. Transcription of a gene by RNA polymerase can be regulated by several mechanisms. Specificity factors alter the specificity of RNA polymerase for a given promoter or set of promoters, making it more or less likely to bind to them (i.e., sigma factors used in prokaryotic transcription). Repressors bind to the Operator, coding sequences on the DNA strand that are close to or overlapping the promoter region, impeding RNA polymerase's progress along the strand, thus impeding the expression of the gene. The image to the right demonstrates regulation by a repressor in the lac operon. General transcription factors position RNA polymerase at the start of a protein-coding sequence and then release the polymerase to transcribe the mRNA. Activators enhance the interaction between RNA polymerase and a particular promoter, encouraging the expression of the gene. Activators do this by increasing the attraction of RNA polymerase for the promoter, through interactions with subunits of the RNA polymerase or indirectly by changing the structure of the DNA. Enhancers are sites on the DNA helix that are bound by activators in order to loop the DNA bringing a specific promoter to the initiation complex. Enhancers are much more common in eukaryotes than prokaryotes, where only a few examples exist (to date).[4]Silencers are regions of DNA sequences that, when bound by particular transcription factors, can silence expression of the gene.[citation needed]
Regulation by RNA
RNA can be an important regulator of gene activity, e.g. by microRNA (miRNA), antisense-RNA, or long non-coding RNA (lncRNA). LncRNAs differ from mRNAs in the sense that they have specified subcellular locations and functions. They were first discovered to be located in the nucleus and chromatin, and the localizations and functions are highly diverse now. Some still reside in chromatin where they interact with proteins. While this lncRNA ultimately affects gene expression in neuronal disorders such as Parkinson, Huntington, and Alzheimer disease, others, such as, PNCTR(pyrimidine-rich non-coding transcriptors), play a role in lung cancer. Given their role in disease, lncRNAs are potential biomarkers and may be useful targets for drugs or gene therapy, although there are no approved drugs that target lncRNAs yet. The number of lncRNAs in the human genome remains poorly defined, but some estimates range from 16,000 to 100,000 lnc genes.[5]
Epigenetic gene regulation
Overview of Epigenetic mechanisms.
Epigenetics refers to the modification of genes that is not changing the DNA or RNA sequence. Epigenetic modifications are also a key factor in influencing gene expression. They occur on genomic DNA and histones and their chemical modifications regulate gene expression in a more efficient manner. There are several modifications of DNA (usually methylation) and more than 100 modifications of RNA in mammalian cells." Those modifications result in altered protein binding to DNA and a change in RNA stability and translation efficiency.[6]
In vertebrates, the majority of gene promoters contain a CpG island with numerous CpG sites.[7] When many of a gene's promoter CpG sites are methylated the gene becomes silenced.[8] Colorectal cancers typically have 3 to 6 driver mutations and 33 to 66 hitchhiker or passenger mutations.[9] However, transcriptional silencing may be of more importance than mutation in causing progression to cancer. For example, in colorectal cancers about 600 to 800 genes are transcriptionally silenced by CpG island methylation (see regulation of transcription in cancer). Transcriptional repression in cancer can also occur by other epigenetic mechanisms, such as altered expression of microRNAs.[10] In breast cancer, transcriptional repression of BRCA1 may occur more frequently by over-expressed microRNA-182 than by hypermethylation of the BRCA1 promoter (see Low expression of BRCA1 in breast and ovarian cancers).[citation needed]
Regulation of transcription in addiction
One of the cardinal features of addiction is its persistence. The persistent behavioral changes appear to be due to long-lasting changes, resulting from epigenetic alterations affecting gene expression, within particular regions of the brain.[11] Drugs of abuse cause three types of epigenetic alteration in the brain. These are (1) histone acetylations and histone methylations, (2) DNA methylation at CpG sites, and (3) epigenetic downregulation or upregulation of microRNAs.[11][12] (See Epigenetics of cocaine addiction for some details.)
Chronic nicotine intake in mice alters brain cell epigenetic control of gene expression through acetylation of histones. This increases expression in the brain of the protein FosB, important in addiction.[13] Cigarette addiction was also studied in about 16,000 humans, including never smokers, current smokers, and those who had quit smoking for up to 30 years.[14] In blood cells, more than 18,000 CpG sites (of the roughly 450,000 analyzed CpG sites in the genome) had frequently altered methylation among current smokers. These CpG sites occurred in over 7,000 genes, or roughly a third of known human genes. The majority of the differentially methylated CpG sites returned to the level of never-smokers within five years of smoking cessation. However, 2,568 CpGs among 942 genes remained differentially methylated in former versus never smokers. Such remaining epigenetic changes can be viewed as "molecular scars"[12] that may affect gene expression.
In rodent models, drugs of abuse, including cocaine,[15] methamphetamine,[16][17] alcohol[18] and tobacco smoke products,[19] all cause DNA damage in the brain. During repair of DNA damages some individual repair events can alter the methylation of DNA and/or the acetylations or methylations of histones at the sites of damage, and thus can contribute to leaving an epigenetic scar on chromatin.[20]
Such epigenetic scars likely contribute to the persistent epigenetic changes found in addiction.[citation needed]
Regulation of transcription in learning and memory
DNA methylation is the addition of a methyl group to the DNA that happens at cytosine. The image shows a cytosine single ring base and a methyl group added on to the 5 carbon. In mammals, DNA methylation occurs almost exclusively at a cytosine that is followed by a guanine.
In mammals, methylation of cytosine (see Figure) in DNA is a major regulatory mediator. Methylated cytosines primarily occur in dinucleotide sequences where cytosine is followed by a guanine, a CpG site. The total number of CpG sites in the human genome is approximately 28 million.[21] and generally about 70% of all CpG sites have a methylated cytosine.[22]
The identified areas of the human brain are involved in memory formation.
In a rat, a painful learning experience, contextual fear conditioning, can result in a life-long fearful memory after a single training event.[23] Cytosine methylation is altered in the promoter regions of about 9.17% of all genes in the hippocampus neuron DNA of a rat that has been subjected to a brief fear conditioning experience.[24] The hippocampus is where new memories are initially stored.[citation needed]
Methylation of CpGs in a promoter region of a gene represses transcription[25] while methylation of CpGs in the body of a gene increases expression.[26]TET enzymes play a central role in demethylation of methylated cytosines. Demethylation of CpGs in a gene promoter by TET enzyme activity increases transcription of the gene.[27]
When contextual fear conditioning is applied to a rat, more than 5,000 differentially methylated regions (DMRs) (of 500 nucleotides each) occur in the rat hippocampus neural genome both one hour and 24 hours after the conditioning in the hippocampus.[24] This causes about 500 genes to be up-regulated (often due to demethylation of CpG sites in a promoter region) and about 1,000 genes to be down-regulated (often due to newly formed 5-methylcytosine at CpG sites in a promoter region). The pattern of induced and repressed genes within neurons appears to provide a molecular basis for forming the first transient memory of this training event in the hippocampus of the rat brain.[24]
After the DNA is transcribed and mRNA is formed, there must be some sort of regulation on how much the mRNA is translated into proteins. Cells do this by modulating the capping, splicing, addition of a Poly(A) Tail, the sequence-specific nuclear export rates, and, in several contexts, sequestration of the RNA transcript. These processes occur in eukaryotes but not in prokaryotes. This modulation is a result of a protein or transcript that, in turn, is regulated and may have an affinity for certain sequences.[citation needed]
Three prime untranslated regions (3'-UTRs) of messenger RNAs (mRNAs) often contain regulatory sequences that post-transcriptionally influence gene expression.[28] Such 3'-UTRs often contain both binding sites for microRNAs (miRNAs) as well as for regulatory proteins. By binding to specific sites within the 3'-UTR, miRNAs can decrease gene expression of various mRNAs by either inhibiting translation or directly causing degradation of the transcript. The 3'-UTR also may have silencer regions that bind repressor proteins that inhibit the expression of a mRNA.[citation needed]
The 3'-UTR often contains miRNA response elements (MREs). MREs are sequences to which miRNAs bind. These are prevalent motifs within 3'-UTRs. Among all regulatory motifs within the 3'-UTRs (e.g. including silencer regions), MREs make up about half of the motifs.[citation needed]
As of 2014, the miRBase web site,[29] an archive of miRNA sequences and annotations, listed 28,645 entries in 233 biologic species. Of these, 1,881 miRNAs were in annotated human miRNA loci. miRNAs were predicted to have an average of about four hundred target mRNAs (affecting expression of several hundred genes).[30] Freidman et al.[30] estimate that >45,000 miRNA target sites within human mRNA 3'-UTRs are conserved above background levels, and >60% of human protein-coding genes have been under selective pressure to maintain pairing to miRNAs.[citation needed]
Direct experiments show that a single miRNA can reduce the stability of hundreds of unique mRNAs.[31] Other experiments show that a single miRNA may repress the production of hundreds of proteins, but that this repression often is relatively mild (less than 2-fold).[32][33]
The effects of miRNA dysregulation of gene expression seem to be important in cancer.[34] For instance, in gastrointestinal cancers, a 2015 paper identified nine miRNAs as epigenetically altered and effective in down-regulating DNA repair enzymes.[35]
The translation of mRNA can also be controlled by a number of mechanisms, mostly at the level of initiation. Recruitment of the small ribosomal subunit can indeed be modulated by mRNA secondary structure, antisense RNA binding, or protein binding. In both prokaryotes and eukaryotes, a large number of RNA binding proteins exist, which often are directed to their target sequence by the secondary structure of the transcript, which may change depending on certain conditions, such as temperature or presence of a ligand (aptamer). Some transcripts act as ribozymes and self-regulate their expression.[citation needed]
Examples of gene regulation
Enzyme induction is a process in which a molecule (e.g., a drug) induces (i.e., initiates or enhances) the expression of an enzyme.[citation needed]
Viruses, despite having only a few genes, possess mechanisms to regulate their gene expression, typically into an early and late phase, using collinear systems regulated by anti-terminators (lambda phage) or splicing modulators (HIV).[citation needed]
Gal4 is a transcriptional activator that controls the expression of GAL1, GAL7, and GAL10 (all of which code for the metabolic of galactose in yeast). The GAL4/UAS system has been used in a variety of organisms across various phyla to study gene expression.[39]
A large number of studied regulatory systems come from developmental biology. Examples include:
The colinearity of the Hox gene cluster with their nested antero-posterior patterning[citation needed]
Pattern generation of the hand (digits - interdigits): the gradient of sonic hedgehog (secreted inducing factor) from the zone of polarizing activity in the limb, which creates a gradient of active Gli3, which activates Gremlin, which inhibits BMPs also secreted in the limb, results in the formation of an alternating pattern of activity as a result of this reaction–diffusion system.[citation needed]
Somitogenesis is the creation of segments (somites) from a uniform tissue (Pre-somitic Mesoderm). They are formed sequentially from anterior to posterior. This is achieved in amniotes possibly by means of two opposing gradients, Retinoic acid in the anterior (wavefront) and Wnt and Fgf in the posterior, coupled to an oscillating pattern (segmentation clock) composed of FGF + Notch and Wnt in antiphase.[40]
Sex determination in the soma of a Drosophila requires the sensing of the ratio of autosomal genes to sex chromosome-encoded genes, which results in the production of sexless splicing factor in females, resulting in the female isoform of doublesex.[41]
Up-regulation is a process which occurs within a cell triggered by a signal (originating internal or external to the cell), which results in increased expression of one or more genes and as a result the proteins encoded by those genes. Conversely, down-regulation is a process resulting in decreased gene and corresponding protein expression.[citation needed]
Up-regulation occurs, for example, when a cell is deficient in some kind of receptor. In this case, more receptor protein is synthesized and transported to the membrane of the cell and, thus, the sensitivity of the cell is brought back to normal, reestablishing homeostasis.
Down-regulation occurs, for example, when a cell is overstimulated by a neurotransmitter, hormone, or drug for a prolonged period of time, and the expression of the receptor protein is decreased in order to protect the cell (see also tachyphylaxis).
Inducible vs. repressible systems
Gene regulation works using operators and repressors in bacteria.
Gene Regulation can be summarized by the response of the respective system:
Inducible systems - An inducible system is off unless there is the presence of some molecule (called an inducer) that allows for gene expression. The molecule is said to "induce expression". The manner by which this happens is dependent on the control mechanisms as well as differences between prokaryotic and eukaryotic cells.
Repressible systems - A repressible system is on except in the presence of some molecule (called a corepressor) that suppresses gene expression. The molecule is said to "repress expression". The manner by which this happens is dependent on the control mechanisms as well as differences between prokaryotic and eukaryotic cells.
The GAL4/UAS system is an example of both an inducible and repressible system. Gal4 binds an upstream activation sequence (UAS) to activate the transcription of the GAL1/GAL7/GAL10 cassette. On the other hand, a MIG1 response to the presence of glucose can inhibit GAL4 and therefore stop the expression of the GAL1/GAL7/GAL10 cassette.[42]
Theoretical circuits
Repressor/Inducer: an activation of a sensor results in the change of expression of a gene[citation needed]
negative feedback: the gene product downregulates its own production directly or indirectly, which can result in[citation needed]
keeping transcript levels constant/proportional to a factor
inhibition of run-away reactions when coupled with a positive feedback loop
creating an oscillator by taking advantage in the time delay of transcription and translation, given that the mRNA and protein half-life is shorter
positive feedback: the gene product upregulates its own production directly or indirectly, which can result in[citation needed]
signal amplification
bistable switches when two genes inhibit each other and both have positive feedback
pattern generation
Study methods
Schematic karyogram of a human, showing an overview of the human genome on G banding, which is a method that includes Giemsa staining, wherein the lighter staining regions are generally more transcriptionally active, whereas darker regions are more inactive.
In general, most experiments investigating differential expression used whole cell extracts of RNA, called steady-state levels, to determine which genes changed and by how much. These are, however, not informative of where the regulation has occurred and may mask conflicting regulatory processes (see post-transcriptional regulation), but it is still the most commonly analysed (quantitative PCR and DNA microarray).[citation needed]
When studying gene expression, there are several methods to look at the various stages. In eukaryotes these include:
Due to post-transcriptional regulation, transcription rates and total RNA levels differ significantly. To measure the transcription rates nuclear run-on assays can be done and newer high-throughput methods are being developed, using thiol labelling instead of radioactivity.[43]
Only 5% of the RNA polymerised in the nucleus exits,[44] and not only introns, abortive products, and non-sense transcripts are degradated. Therefore, the differences in nuclear and cytoplasmic levels can be seen by separating the two fractions by gentle lysis.[45]
Alternative splicing can be analysed with a splicing array or with a tiling array (see DNA microarray).[citation needed]
All in vivo RNA is complexed as RNPs. The quantity of transcripts bound to specific protein can be also analysed by RIP-Chip. For example, DCP2 will give an indication of sequestered protein; ribosome-bound gives and indication of transcripts active in transcription (although a more dated method, called polysome fractionation, is still popular in some labs)[citation needed]
↑de Souza MF, Gonçales TA, Steinmetz A, Moura DJ, Saffi J, Gomez R, Barros HM (April 2014). "Cocaine induces DNA damage in distinct brain areas of female rats under different hormonal conditions". Clinical and Experimental Pharmacology & Physiology. 41 (4): 265–9. doi:10.1111/1440-1681.12218. PMID24552452. S2CID20849951.
↑Johnson Z, Venters J, Guarraci FA, Zewail-Foote M (June 2015). "Methamphetamine induces DNA damage in specific regions of the female rat brain". Clinical and Experimental Pharmacology & Physiology. 42 (6): 570–5. doi:10.1111/1440-1681.12404. PMID25867833. S2CID24182756.
↑Weber M, Hellmann I, Stadler MB, Ramos L, Pääbo S, Rebhan M, Schübeler D (April 2007). "Distribution, silencing potential and evolutionary impact of promoter DNA methylation in the human genome". Nat. Genet. 39 (4): 457–66. doi:10.1038/ng1990. PMID17334365. S2CID22446734.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.