
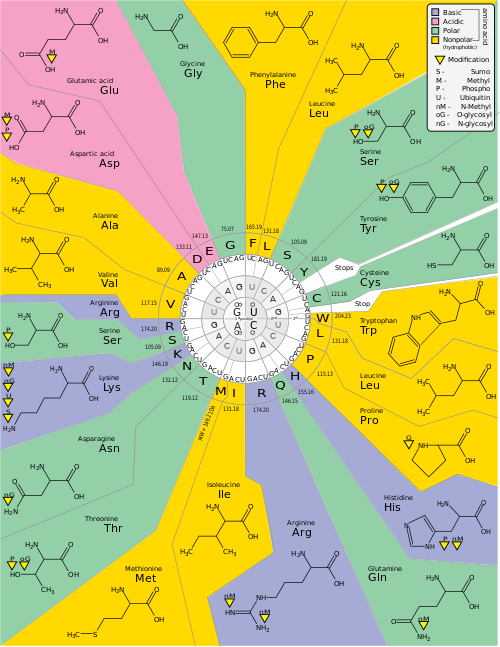
Genetic code is a set of rules used by living cells to translate information encoded within genetic material (DNA or RNA sequences of nucleotide triplets or codons) into proteins. Translation is accomplished by the ribosome, which links proteinogenic amino acids in an order specified by messenger RNA (mRNA), using transfer RNA (tRNA) molecules to carry amino acids and to read the mRNA three nucleotides at a time. The genetic code is highly similar among all organisms and can be expressed in a simple table with 64 entries.
Contents
- History
- Codons
- Expanded genetic codes (synthetic biology)
- Features
- Reading frame
- Start and stop codons
- Effect of mutations
- Degeneracy
- Codon usage bias
- Alternative genetic codes
- Non-standard amino acids
- Variations
- Inference
- Origin
- See also
- References
- Further reading
- External links
The codons specify which amino acid will be added next during protein biosynthesis. With some exceptions, [1] a three-nucleotide codon in a nucleic acid sequence specifies a single amino acid. The vast majority of genes are encoded with a single scheme (see the RNA codon table). That scheme is often called the canonical or standard genetic code, or simply the genetic code, though variant codes (such as in mitochondria) exist.




{kind=link}